ÁREA: Iniciação Científica
TÍTULO: INVESTIGAÇÃO COMPUTACIONAL DO MECANISMO DE REAÇÕES DE ELIMINAÇÃO
AUTORES: DE BRITO, H.G. (U.F.PA) ; ALMEIDA, R.C.O. (U.F.PA) ; DOS SANTOS, M.A.B. (U.F.PA) ; MARTINS, E.A. (U.F.PA) ; SANTOS, C.B.R. (U.F.PA) ; LOBATO, M.S. (U.F.PA) ; MACIEL, A.A. (U.F.PA) ; FIGUEIREDO, A.F. (U.F.PA) ; MACÊDO, W.J.C. (U.F.PA) ; BARBOSA, J.P. (U.F.PA) ; PINHEIRO, J.C. (U.F.PA)
RESUMO: RESUMO: Embora reação de eliminação total seja um processo relativamente simples, o mecanismo é complexo e envolve quatro diferentes mudanças de ligação: (1) Formação de uma nova ligação Y-H; (2) Clivagem da ligação C-H;(3) Formação da ligação C-C e (4) Clivagem da ligação C-H. O entendimento da estrutura eletrônica dessas reações foi feito por métodos computacionais em diversos níveis de teoria e a construção de mapas de potencial eletrostático foi utilizada como ferramenta interpretativa da desidroalogenação de haletos de alquila.
PALAVRAS CHAVES: palavras chaves: reação de eliminação; química computacional; potencial eletrostático molecular.
INTRODUÇÃO: INTRODUÇÃO: A química computacional na atualidade é uma ferramenta cada vez mais útil e desejável para o ensino e pesquisa, para estudantes tanto do ensino médio quanto superior. No ensino de química orgânica é possível obter resultados altamente confiáveis de cálculos de propriedades, sendo aplicada com sucesso no estudo de uma ampla faixa de problemas de interesse químico, tais como: cinética de reações (estado de transição), reatividade e estabilidade conformacional (Hessley,2000). Este trabalho tem como objetivos: estudar reações de desidroalogenação de haletos alquila com auxílio de cálculos computacionais; interpretação dos intermediários de reações por mapas de potencial eletrostático (Murray,1996); Motivar os estudantes a investigar os conceitos de química orgânica através de modelagem molecular, visto que, existe uma diversidade de programas e bancos de dados disponíveis em rede.
MATERIAL E MÉTODOS: MATERIAL E MÉTODOS: A proposta foi desenvolvida investigando três diferentes tipos de reações de eliminação. Procurou-se, inicialmente, uma reação simples na qual se obtivesse um único produto. Nas demais reações utilizou-se um único substrato e diferentes reagentes nucleofílicos querendo investigar o efeito de grupos volumosos, a reatividade e estabilidade conformacional.
Os substratos 2-bromo-2-metil-propano e 2-bromo-2-metil-butano e os reagentes etoxi e terbutoxi foram construídos com auxílio do programa GaussView 1.0 e otimizadas (obtendo as estruturas de mínima energia) com cálculos semi-empíricos (AM1 e PM3), ab initio (HF/3-21G*) e DFT (B3LYP/3-21G*) utilizando PCs operando em sistema GNU/Linux. A energia total, dos orbitais HOMO e LUMO, do estado de transição e calor de formação foram calculados no programa Gaussian 98 para os métodos citados anteriormente.
O programa MOLEKEL foi utilizado para visualização do potencial eletrostático molecular, que representa uma superfície de contorno obtida a partir das cargas de Mullikan, sendo então possível realizar movimentos tridimensionais das estruturas, possibilitando ao estudante a observação de uma estrutura microscópica através de um modelo macroscópico (Plükiger, 2000-2001).
RESULTADOS E DISCUSSÃO: RESULTADOS E DISCUSSÃO: Para a desidroalogenação de haleto de alquila, a teoria do funcional densidade foi o método que melhor descreveu as propriedades estruturais e eletrônicas dos compostos estudados. Os valores dos calores de formação e a construção das estruturas dos estados de transição obtidos para cada reação possibilitou o melhor entendimento do mecanismo das reações de eliminação. Para a reação entre o brometo de t-butila e o íon alcóxido observou-se a formação de um só estado de transição (TS) com Hf em (-1824,395 Kcal/mol). A reação entre o 2-bromo-2-metil-butano e o íon alcóxido possibilita a formação de dois intermediários de reação TS1 e TS2. TS1 é 7,82 Kcal/mol mais estável que TS2 formando, portanto, o 2-metil-2-buteno 2,48.10-3 Kcal/mol mais estável que 2-metil-1-buteno. A Figura 1 mostra o mapa de potencial eletrostático molecular para a reação de formação do 2-metil-2-buteno.
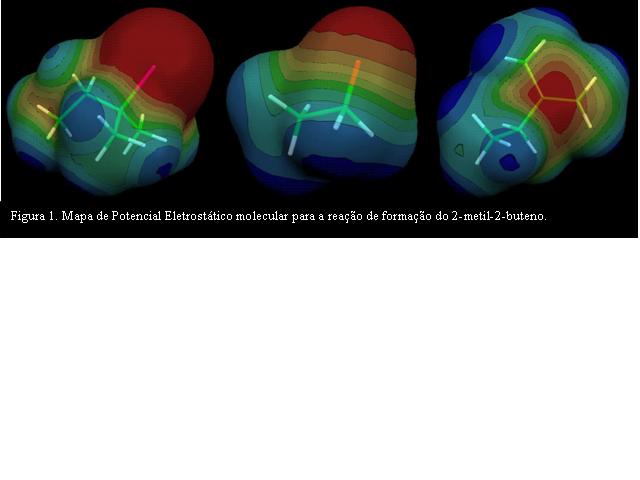
CONCLUSÕES: CONCLUSÕES: O uso de modelagem computacional deve ser incorporado nas grades curriculares, pois possibilita abordar um número maior de tópicos e um aprofundamento da teoria. Na primeira reação estudada é predito e observado que o 2metil-propeno é o composto mais estável. A investigação computacional do estado de transição da reação entre 2-metil-2-bromobutano com íon alcóxido é possível predizer que o alqueno mais substituído é o composto majoritário. No MEP a região de potencial eletrostática negativa (região vermelha) enfatiza que a dupla ligação formada concentra-se no centro da molécula.
AGRADECIMENTOS: AGRADECIMENTOS: Ao LQTC- Laboratório de Química Teórica e Computacional pela infra-estrutura.
REFERÊNCIAS BIBLIOGRÁFICA: REFERÊNCIAS:
Hessley, R. K. J. Chem. Educ. 2000, 77, 794.
Murray, J. S.; Sen, K. Molecular Eletrostatic Potentials: Concepts and Applications, Elsevier, 1996, Amsterdam.
Plükiger, P.; Lüthi, H. P.; Portmann, S.; Webber, J.; MOLEKEL 4.1, Swiss Center for Scientific Computing, Switzerland, 2000-2001.
