ISBN 978-85-85905-10-1
Área
Química Orgânica
Autores
Silva, A.I.S. (UFPE) ; Junior, G.C. (UFPE) ; Gonçalves, S.M.C. (UFPE)
Resumo
Neste trabalho realizamos um estudo teórico e computacional das propriedades moleculares de três diferentes pares de ésteres diastereotópicos obtidos a partir de reações envolvendo um agente de derivatização quiral, um álcool, com três ácidos carboxílicos quirais racêmicos. Utilizamos o nível de cálculo DFT B3LYP/6- 31++G(d,p) e o programa GAUSSIAN 2009. As geometrias dos ésteres diastereotópicos formados de cada reação, foram otimizadas para os diferentes pares e correspondem a pontos de mínimo na curva de energia potencial. As estabilidades energéticas dos pares de ésteres diastereotópicos são similares, indicando que ambas as estruturas devem ser igualmente formadas. Por fim, os valores do momento de dipolo dos compostos estudados são consideravelmente distintos.
Palavras chaves
Cálculos Teóricos; Excesso Enantiomérico; ADQ
Introdução
Os enantiômeros podem ter funções diferenciadas no nosso organismo como por exemplo, os medicamentos varfarina e o propanolol. O enantiômero (S)-varfarina é um anticoagulante seis vezes mais potente que seu respectivo enantiômero R e o (S)-propanolol é um anti-hipertensivo e anti-arrítmico usado no tratamento de doenças do coração enquanto que o enantiômero R atua como um anticoncepcional (PARKER, 1991). Através da espectroscopia de Ressonância Magnética Nuclear (RMN) é possível utilizar estratégias para determinar a pureza enantiomérica pela reação de uma substância conhecida, Agente de Derivatização Quiral (ADQ), com os substratos quirais racêmicos, onde a reação destas moléculas (esterificação) possui o núcleo de selênio inserido nesse ADQ. É extremamente importante que ambas as estruturas dos diferentes enantiômeros possam ter a mesma estabilidade energética para reagir com o ADQ, a fim de obter duas estruturas diastereotópicas na mesma proporção (SILVA and GONÇALVES, 2013). Neste trabalho temos como objetivo o estudo teórico e computacional das propriedades moleculares de três diferentes pares de ésteres diastereotópicos obtidos a partir de reações envolvendo o ADQ, o álcool (R)-3-fenil-2- (fenilselanil)propanol, e três ácidos carboxílicos quirais racêmicos: ácido 2-bromo-butanóico, ácido 2-bromo-3-metil-butanóico e o ácido 2-fenil- butanóico.
Material e métodos
Foram realizados cálculos usando a Teoria do Funcional de Densidade (DFT) (BECKE, 1993) com o funcional B3LYP (GEERLINGS et al, 2003) com o conjunto de funções base 6-31++G(d,p) para otimização completa de geometria e cálculo dos modos vibracionais dos complexos. Todos os cálculos foram realizados utilizando-se o programa de química quântica computacional GAUSSIAN 2009 (FRISCH et al, 2009).
Resultado e discussão
As geometrias otimizadas dos três diferentes pares de ésteres diastereotópicos
são apresentadas na figura 1. A partir da otimização destas geometrias
verificamos que não foi obtido nenhum modo vibracional imaginário, ou seja,
todas as estruturas estudadas neste trabalho são correspondentes a pontos de
mínimo na curva de energia potencial.
Em seguida, passamos a analisar a diferença de energia, ΔE, para cada par de
ésteres diastereotópicos, ΔE, neste processo adicionamos a correção da energia
do ponto zero, obtendo assim a variação de energia acrescida desta correção,
ΔEZPE. Observamos a partir dos nossos resultados que praticamente não há
diferença energética entre os compostos estudados, isto pode ser explicado
porque as pequenas diferenças de conformação não chegaram a afetar a
estabilidade do sistema, uma vez que os sítios quirais estão a uma distância de
quatro ligações. Estes resultados indicam que ambas as estruturas dos ésteres
diastereotópicos (RR e RS) devem ser formadas na mesma proporção (figura 1).
Posteriormente, passamos a investigar os valores de momento de dipolo, μ, dos
compostos. Nesta etapa verificamos que há uma diferença substancial entre os
pares de ésteres diastereotópicos, onde observamos, por exemplo, que para um
deles, o 2-bromo-3-metilbutanoato de 3-fenil-2-(fenilselanil)propila, há uma
diferença de momento de dipolo, Δμ, de 2,33 D. Estes resultados mostram que
embora as estabilidades energéticas sejam similares as propriedades físicas dos
compostos são bem distintas entre si.
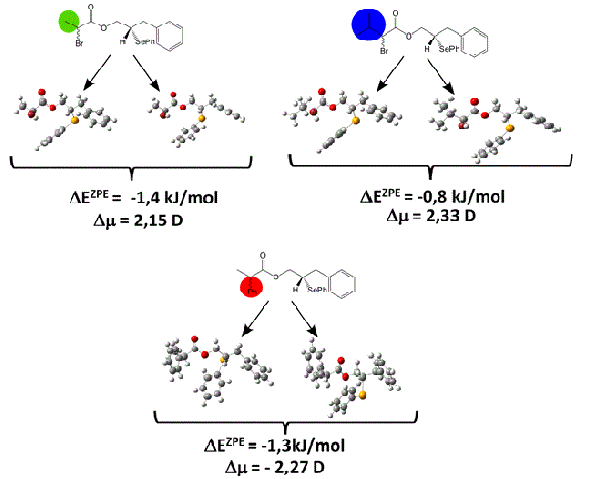
Geometrias otimizadas dos ésteres diastereotópicos estudados e as correspondentes diferenças de energia e de momento de dipolo.
Conclusões
As geometrias otimizadas dos diferentes pares de ésteres diastereotópicos correspondem a pontos de mínimo na curva de energia potencial. As estabilidades energéticas dos pares de ésteres diastereotópicos são similares, indicando que ambas as estruturas devem ser formadas em proporções iguais. Por fim, os valores de momento de dipolo dos compostos estudados são distintos.
Agradecimentos
PRONEX, FACEPE, PROPESQ/UFPE, CNPq, DQF, UFPE.
Referências
1. PARKER, D.; NMR Determination of Enantiomeric Purity. Chemial Review, 91, p. 1441, 1991;
2. SILVA, A. I. S.; GONÇALVES, S. M. C.; Síntese do (R)-3-fenil-2-(fenilselanil)propanol como Auxiliar Quiral para Determinação de Excesso Enantiomérico de Ácidos Carboxílicos Quirais por Ressonância Magnética Nuclear de 77Se. Monografia de Graduação, DQF/UFPE, 2013;
3. BECKE, A. D.; Density-Functional Thermochemistry. III. The Role of Exact Exchange. The Journal of Chemical Physics, vol. 98, p. 5648, 1993;
4. GEERLINGS, P.; PROFT, F.; LANGENAEKER, W.; Conceptual Density Functional Theory. Chemical Reviews, vol. 103, p. 1793-1874, 2003;
5. FRISCH, M. J. et al; Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT, 2009.
Patrocinadores





Apoio





Realização
