ISBN 978-85-85905-10-1
Área
Química Orgânica
Autores
Machado, C.M.B. (UFPE) ; Moura, G.L.C. (UFPE)
Resumo
A determinação da configuração absoluta de moléculas orgânicas é um processo muito trabalhoso experimentalmente. Neste trabalho, estudamos métodos DFT, com diferentes funções de base, para a previsão da configuração absoluta de duas moléculas orgânicas, uma rígida e outra com duas conformações. Para a molécula rígida, cálculos com baixo custo computacional foram suficientes para um resultado semelhante ao ângulo experimental. Para a molécula flexível analisada, é necessário um estudo que contemple a possibilidade de formação de dímeros, que interferem no resultado final do ângulo.
Palavras chaves
Quiralidade; Configuração absoluta; Métodos DFT
Introdução
Um dos conceitos mais importantes na química orgânica é o de quiralidade. Isômeros quirais possuem a mesma fórmula molecular e sequência de átomos ligados, porém diferentes arranjos tridimensionais dos átomos. A atribuição da configuração absoluta de uma molécula quiral envolve a determinação do arranjo espacial dos grupos ligados a cada um dos seus carbonos assimétricos. Muitos produtos naturais são quirais e, atualmente, a atribuição da sua configuração absoluta é um processo muito laborioso que inclui a combinação de métodos espectroscópicos e cristalográficos com a síntese total ou parcial da molécula. Um grande diferencial entre isômeros quirais é o seu comportamento frente a um feixe de luz linearmente polarizado. Cada um dos diferentes isômeros ópticos da molécula irá rodar o plano de polarização da luz por um ângulo diferente. Frequentemente, o ângulo de rotação óptica é medido para um único comprimento de onda que, por convenção, é tomado como sendo o comprimento de onda da linha D do átomo de sódio (λ=589nm). Esta medida é chamada de poder rotatório específico [α]D. Recentemente, tem-se observado um crescimento no número de trabalhos que utilizam cálculos computacionais dos valores de [α]D visando ajudar na atribuição da configuração absoluta de moléculas orgânicas. (HEDEGARD et. al. 2012; ZUBER et. al. 2005; EVIDENTE et.al. 2011). O objetivo deste trabalho é o de encontrar um método de química quântica de baixo custo computacional capaz de fornecer valores de [α]D teóricos que possam ser utilizados na atribuição da configuração absoluta de moléculas orgânicas.
Material e métodos
Selecionamos duas moléculas, sendo uma rígida e outra flexível, para os nossos estudos (figura 1). Os cálculos foram realizados, no programa GAUSSIAN 09, utilizando-se três métodos DFT (B3LYP, PBE e PBE0) e dezoito funções de base gaussianas diferentes. Otimizamos as geometrias das moléculas tanto isoladas quanto em solução. O efeito do solvente foi simulado com o modelo PCM. Os ângulos de rotação foram calculados utilizando-se GIAOs e, no caso da molécula flexível, o ângulo final foi obtido através de uma média de Boltzmann utilizando-se energias com correção do ponto zero.
Resultado e discussão
As moléculas escolhidas para o nosso estudo foram o (R,R)-dimetil oxirano, como
molécula rígida, e a (S)(+)4-fenil-2-oxazolidinona, que possui duas conformações
acessíveis. A molécula rígida possui um ângulo de rotação óptica experimental de
58,8o e a molécula flexível um ângulo de 48o em
clorofórmio. As geometrias foram otimizadas por três métodos DFT empregando-se
dezoito funções de base gaussianas diferentes, contemplando bases com custos
computacionais altos e baixos. Com estas geometrias, calculamos os ângulos de
rotação utilizando os mesmos métodos e funções de base.
Os ângulos calculados foram mais sensíveis a mudanças no conjunto de funções de
base do que ao método DFT empregado. Para a molécula rígida isolada, oito das
dezoito funções de base deram bons resultados em comparação com o valor
experimental (os valores variaram de 48,75o a 66,89o).
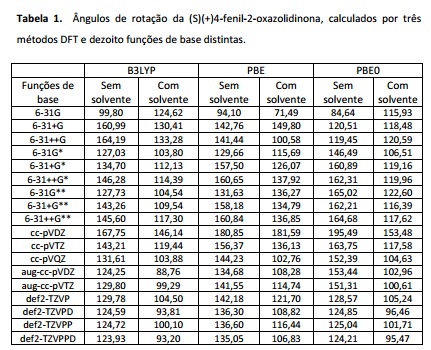
Para a molécula flexível, o ângulo final convergiu para um resultado semelhante,
em torno de 144,7o, em quinze das dezoito funções de base.
Comportamento semelhante foi observado para essa molécula calculada na presença
do solvente, porém com ângulos aproximadamente 30o menores em relação
à molécula isolada, e mais próximo do valor experimental (tabela 1).
No caso da molécula flexível, a discrepância entre os resultados teóricos e
experimentais provavelmente se dá pelo fato dessa molécula poder gerar dímeros,
e esses dímeros certamente influenciam o valor do ângulo de rotação
experimental.
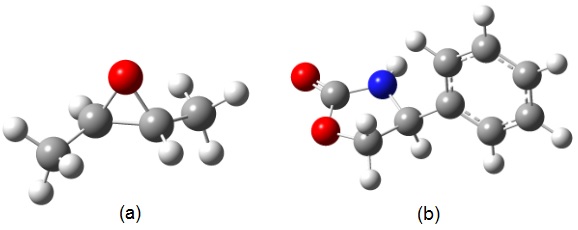
Moléculas estudadas: (a) (R,R)-dimetil oxirano; (b) (S)(+)4-fenil-2-oxazolidinona.
Conclusões
Na molécula rígida foi possível encontrar cinco funções de base de baixo custo computacional capazes de dar resultados semelhantes ao experimental: as bases 6- 31++G*, 6-31+G**, def2-TZVP, def2-TZVPD e def2-TZVPP. Para a molécula flexível, apesar dos resultados convergirem para valores relativamente próximos, mais estudos são necessários levando-se em consideração a capacidade da molécula formar dímeros para uma melhor descrição do ângulo de rotação óptica desta molécula.
Agradecimentos
Os autores agradecem à FACEPE, ao CNPQ e ao PRONEX/FACEPE pelo financiamento deste trabalho.
Referências
HEDEGARD, E. D.; JESEN, F.; KONGSTED, J. Basis Set Recommendations for DFT Calculations of Gas-Phase Optical Rotation at Different Wavelengths. J. Chem. Theory Comput. 8, 4425−4433, 2012.
ZUBER, G.; GOLDSMITH, M.R.; HOPKINS, T. D.; BERATAN, D. N.; WIPF, P. Systematic Assignment of the Configuration of Flexible Natural Products by Spectroscopic and Computational Methods: The Bistramide C Analysis. Organic Letters. Vol 7, no. 23, 5269-5272, 2005.
Evidente, A.; Superchi, S.; Cimmino, A. ; Mazzeo, G.; Mugnai, L.; Rubiales, D. ; Andolfi A.; Fernández, A. M. V. Regiolone and Isosclerone, Two Enantiomeric Phytotoxic Naphthalenone Pentaketides: Computational Assignment of Absolute Configuration and Its Relationship with Phytotoxic Activity. European Journal of Organic Chemistry. Vol 2011, Issue 28, 5564-5570, 2011
Patrocinadores





Apoio





Realização
