ISBN 978-85-85905-10-1
Área
Química Orgânica
Autores
Pinheiro, S.M. (UFPB) ; dos Santos Filho, J.M. (UFPE)
Resumo
Sistemas heterocíclicos fazem parte da estrutura de várias moléculas com vasta aplicação na terapêutica. Estudos detalhados mostram que tais blocos são responsáveis pela atividade biológica ou farmacológica observada. Planejamento empregando a abordagem de hibridação molecular indicou uma combinação promissora entre os grupos 1,2,4-oxadiazol e aciltiossemicarbazida. Em uma proposta mais eficiente, os derivados do fragmento contendo o anel 1,2,4-oxadiazol foram sintetizados em uma única etapa com rendimentos variando de bons a excelentes. A otimização da metodologia permitiu a obtenção dos derivados na escala grama. Os intermediários sintéticos e os produtos finais terão o perfil biológico traçado para a atividade anticancerígena.
Palavras chaves
1,2,4-oxadiazol; atividade biológica; N-acil-hidrazonas
Introdução
A entrada de um novo medicamento no mercado é um processo laborioso, caro e que exige muita sofisticação do ponto de vista tecnológico. Grande parte dos fármacos comercialmente disponíveis no tratamento das mais diversas enfermidades tem incorporado em sua estrutura um bloco heterocíclico. Várias moléculas contendo o anel 1,2,4-oxadiazólico são relatadas como detentoras de efeitos biológicos como anti-asmático, anti-diabético, antiinflamatório, anti-microbial, anti-tumoral, imunossupressor e neuroprotetor.[1] Após um planejamento empregando a abordagem de hibridação molecular, nosso grupo de pesquisa reuniu dois blocos farmacofóricos, o anel 1,2,4-oxadiazol e a aciltiossemicarbazida. Segundo a proposta inicial, os produtos finais foram obtidos em rendimentos globais variando de 12 a 28% acompanhados de um subproduto de ciclização (2- 10%). A formação desses subprodutos foi evitada sintetizando-se derivados contendo espaçadores de carbono entre o anel 1,2,4-oxadiazol e a função hidrazida (Fig. 1). Contudo a metodologia empregada na obtenção dos ésteres precursores dessas hidrazidas apresentavam rendimentos modestos (31-43%). Essa rota sintética foi aprimorada em um processo de construção do anel 1,2,4- oxadiazol empregando um reagente com a função éster pré-existente (Fig. 2). Com esses compostos em mãos, os produtos finais serão obtidos em escala de grama por metodologia já otimizada e submetidos a avaliação da sua atividade anticâncer.
Material e métodos
Os compostos 12a-h foram obtidos a partir da reação, em atmosfera de N2, entre as arilamidoximas 6a-h e o 4-cloro-4-oxo-butirato de etila (11), usando diisopropiletilamina e fluoreto de tributilamônio em THF andiro a 75ºC por 18h. Após remoção do solvente, o resíduo foi solubilizado em CH2Cl2 e lavado com soluções HCl 10% e NaHCO3 saturada. Os produtos brutos foram purificados por cromatografia e devidamente caracterizados. Os ésteres 12a-h foram convertidos às hidrazidas 10a-h a partir do refluxo das suas suspensões em uma solução aquosa de hidrazina 55% por 1h. Após esse período, os produtos foram filtrados a vácuo e após 12h em dessecador, tiveram seus pontos de fusão obtidos. As aciltiossemicarbazidas-1,2,4-oxadiazolpropiônicas 5a-h foram obtidas a partir da reação entre o isotiocianato de fenila em THF a temperatura ambiente por 10 min. Os produtos foram recristalizados em etanol e devidamente caracterizados. A cromatografia em camada fina foi efetuada em placas de alumínio contendo gel de sílica 60 F254. As cromatografias em coluna foram realizadas usando gel de sílica 60. Os solventes usados nas cromatografias foram hexano e acetato de etila puros ou em misturas binárias com gradiente de concentração em ordem crescente de polaridade. A revelação das substâncias nas cromatoplacas ocorreu por irradiação com luz ultravioleta no comprimento de onda de 254nm e por saturação em câmara de I2. Todos os produtos foram caracterizados por infravermelho em aparelho Bruker Tensor27. Os espectros de RMN (1H e 13C) foram obtidos em espectrômetro Varian 300 ou 400MHz. Análises de LCMS (cromatografia líquida acoplada a espectrometria de massa) foram feitas em LCMS-IP-TOF da Shimadzu. Os pontos de fusão foram medidos em fusiômetro digital com variação de ±2ºC.
Resultado e discussão
Trabalhos anteriores do nosso grupo de pesquisa relatam a obtenção de derivados
contendo simultaneamente os grupos farmacofóricos 1,2,4-oxadiazol e
aciltiossemicarbazida. A obtenção desses produtos foi acompanhada da formação de
um produto ciclização espontânea (2-10%), que foi eliminada após a introdução de
um espaçador de 2 átomos de carbono entre o anel 1,2,4-oxadiazol e a função
hidrazida (Fig. 1).[2]
A obtenção dos ésteres 12a-h em uma etapa tornou a rota mais eficiente, além de
atrativa do ponto de vista operacional. Baseando-se no trabalho de Maingot e
colaboradores,[3] a uma solução da arilamidoxima em THF anidro e sob atmosfera
inerte, adicionam-se o 4-cloro-4-oxo-butirato de etila e a Et3N, ambos gota a
gota. Após 1h sob refluxo, introduz-se o catalisador TBAF (10mol%). Após
purificação por cromatografia em coluna, os produtos foram obtidos com
rendimentos que variaram de moderados a excelentes. Ressalta-se que os ésteres
etílicos são de caráter inédito na literatura. Os intermediários 1,2,4-
oxadiazolpropionil-hidrazidas (10a-h) correspondentes ainda não possuem suas
constantes físicas e espectroscópicas publicadas. Seus rendimentos variam entre
62 e 85% (Fig. 2 ).
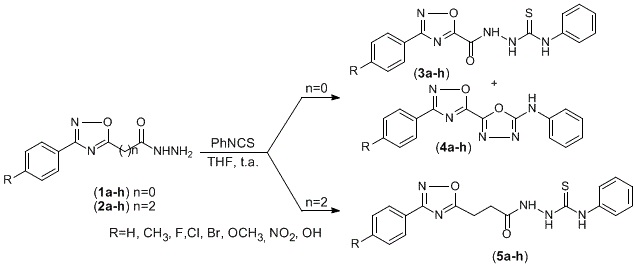
Alternativa para a ciclização espontânea dos derivados 1,2,4-oxadiazol-aciltiossemicarbazida diretamente acoplados.
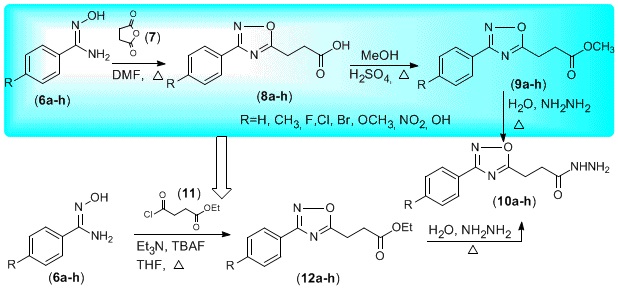
Otimização da rota sintética na obtenção dos ésteres 12a-e e hidrazidas 10a-h.
Conclusões
A metodologia de otimização para a obtenção dos ésteres propiônicos (12a-h) mostrou-se eficiente e confirmou a hipótese de que a ciclização espontânea observada na série 4a-h está associada às características estruturais desses compostos. A rota sintética planejada e descrita neste trabalho foi executada com sucesso, levando a rendimento global maior para as aciltiossemicarbazidas 5a-h.Os compostos obtidos encontram-se sob avaliação da sua atividade anticancerígena.
Agradecimentos
Os autores são gratos à equipe da Central Analítica do Departamento de Química Fundamental da UFPE pelas análises espectroscópicas.
Referências
[1] Bora, R. O.; Dar, B.; Pradhan, V.; Farooqui, M. Mini-Reviews in Medicinal Chemistry 2013, 13, 7, 355.
[2] dos Santos Filho, J. M.; de Lima, J. G.; Leite, L. F. C. C. J. Heterocyclic Chem. 2009, 46, 722.
[3] Maingot, L.; Leroux, F.; Landry, V.; Dumont, J.; Nagase, H.; Villoutreix, B.; Sperandio, O.; Deprez-Poulain, R.; Deprez, B. Bioorg. Med. Chem. Lett. 2010, 20, 6213.
Patrocinadores





Apoio





Realização
