ISBN 978-85-85905-10-1
Área
Iniciação Científica
Autores
Silva, T.G. (UFPE) ; Machado, C.M.B. (UFPE) ; Silva, R.P.G. (UFPE) ; Lima, N.B. (UFPE)
Resumo
Neste trabalho realizamos o estudo teórico e computacional das propriedades termodinâmicas teóricas dos complexos envolvendo o ácido benzoico e compostos orgânicos. As propriedades termodinâmicas foram calculadas com método DFT B3LYP/6- 31++G(d,p) utilizando para isto o programa de química quântica computacional Gaussian 2009. Todas as geometrias dos complexos foram correspondentes a um ponto de mínimo na curva de energia potencial, uma vez que nenhum modo vibracional imaginário foi encontrado. Os resultados mostraram que as entalpias de reação dos complexos envolvendo ácido benzoico e os compostos FEN e DIBIPI foram as mais negativas indicando que estes complexos são os mais estáveis energeticamente dentre o grupo de complexos estudados.
Palavras chaves
Ligações de hidrogênio; propriedades termodinâmic; cálculos DFT
Introdução
Complexos de ligação de hidrogênio são formados a partir da interação entre um hidrogênio ácido de moléculas conhecidas como “doadoras de próton” com regiões de alta densidade eletrônica, a exemplo de pares de elétrons isolados de átomos como O, F, N(NADVORNY et al, 2011 e DA SILVA et al, 2005) ou de elétrons pi em ligações duplas (C=C) e triplas (C≡C) (BELARMINO et al, 2013) de moléculas conhecidas como “aceitadoras de próton”. Estas ligações são energeticamente mais fracas quando comparadas a ligações covalentes e iônicas, mas a sua presença em sistemas orgânicos ou inorgânicos contribui para diversas propriedades, como por exemplo, aumento na estabilidade energética. Neste trabalho temos como objetivo o estudo teórico e computacional das propriedades termodinâmicas teóricas dos complexos envolvendo o ácido benzoico, o qual age como molécula doadora de próton, com vários compostos orgânicos, figura 1, os quais por sua vez podem ser considerados como pré-ligantes de complexos de íons lantanídeos trivalentes, como por exemplo o európio, uma vez que os grupos Y=O, Y=C ou S e N, se coordenam facilmente a estes metais de transição interna.
Material e métodos
As propriedades termodinâmicas teóricas e a otimização completa de geometria dos complexos foram calculadas com no nível de cálculo DFTB3LYP/6-31++G(d,p) (BECKE et al, 1993 e GEERLINGS et al, 2003) utilizando para isto o programa de química quântica computacional Gaussian 2009 (FRISCH et al, 2009).
Resultado e discussão
Consideramos neste trabalho o complexo entre duas moléculas de ácido benzoico,
em que há apenas uma ligação de hidrogênio ao invés do dímero ácido, no sentido
de verificarmos a força da ligação de hidrogênio em situação similar à dos
demais complexos. Caso considerássemos o dímero ácido, o valor da entalpia de
reação dos demais complexos deveria ser multiplicada por um fator de dois porque
a reação em questão seria: dímero ácido benzoico + 2 pré-ligantes→ 2 complexos
pré-ligante∙∙∙ácido benzoico. A figura 2 apresenta as geometrias completamente
otimizadas dos complexos entre ácido benzoico e o grupo de pré-ligantes
escolhidos neste trabalho. Todas as geometrias dos complexos foram
correspondentes a um ponto de mínimo na curva de energia potencial, uma vez que
nenhum modo vibracional imaginário foi encontrado.
Também apresentamos nesta figura os valores das entalpias teóricas de reação,
ΔHr, as quais foram calculadas da seguinte maneira: ΔHr = Hf complexo pré-
ligante∙∙∙ácido benzoico - Hf pré-ligante - Hf ácido benzoico.
Analisando os nossos resultados temos que os complexos formados pelos pré-
ligantes orgânicos DIBIPI e FEN possuem os valores de ΔHr mais negativos, isto
significa dizer que estes pré-ligantes provavelmente realizam as ligações de
hidrogênio mais fortes quando comparado com as ligações de hidrogênio dos demais
pré-ligantes estudados neste trabalho, por sua vez quando consideramos o
complexo formado por dois pré-ligantes ácido benzoico, o valor de ΔHr é o menos
negativo, isto indica que provavelmente este complexo é o mais fraco
energeticamente.
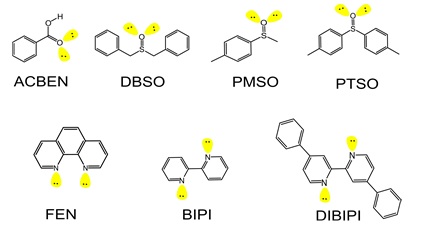
Estrutura dos pré-ligantes ACBEN, DBSO, PMSO, FEN, BIPI e DIBIPI.
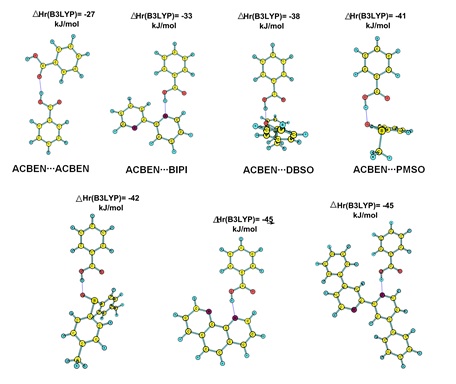
Geometria otimizada dos complexos de ligação de hidrogênio pré-ligante∙∙∙ácido benzoico e valores da entalpia de reação.
Conclusões
Neste trabalho estudamos as propriedades termodinâmicas teóricas de complexos entre o ácido benzoico e pré-ligantes orgânicos. Todas as geometrias dos complexos foram correspondentes a um ponto de mínimo na curva de energia potencial, uma vez que nenhum modo vibracional imaginário foi encontrado. Os resultados mostraram que as entalpias de reação dos complexos envolvendo ácido benzoico e os pré-ligantes FEN e DIBIPI foram as mais negativas indicando que estes complexos são os mais estáveis dentre o grupo de complexos estudados.
Agradecimentos
Os autores agradecem ao PET/CAPES, à FACEPE, ao CNPq, à PROAES/UFPE e ao PRONEX/FACEPE.
Referências
NADVORNY, D. AND JOÃO B. P. DA SILVA, Hydrogen Bond Complexes of Hydantoin : A Theoretical Study. , Int. J. Quantum Chem., 111, 1436–1443, 2011.
DA SILVA, J. B. P.; SILVA JÚNIOR, M. R.; RAMOS, M. N.; GALEMBECK, S. E., An Ab Initio Study of the Electronic and Vibrational Properties of pyrazine⋯HX and XH⋯pyrazine⋯HX Hydrogen-Bonded Complexes (X=F, NC, Cl, CN and CCH), J. Mol. Struct. 744-747, 217–220, 2005.
BELARMINO, M. K. D. L.; LIMA, N. B. D.; RAMOS, M. N., Hydrogen Bonds between Acetylene and Formic Acid: An Ab Initio Study, Int. J. Quantum Chem. 112, 3246–3251, 2012.
A.D. BECKE, Density‐functional thermochemistry. III. The role of exact Exchange. The Journal of Chemical Physics, Vol. 98, P. 5648, 1993.
P. GEERLINGS, F. DE PROFT, W. LANGENAEKER, Conceptual Density Functional Theory. Chemical Reviews. Vol. 103, P. 1793–1874, 2003.
M. J. FRISCH, G. W. TRUCKS, H. B. SCHLEGEL, G. E. SCUSERIA, M. A. ROBB, J. R. CHEESEMAN, G. SCALMANI, V. BARONE, B. MENNUCCI, G. A. PETERSSON, H. NAKATSUJI, M. CARICATO, X. LI, H. P. HRATCHIAN, A. F. IZMAYLOV, J. BLOINO, G. ZHENG, J. L. SONNENBERG, M. HADA, M. EHARA, K. TOYOTA, R. FUKUDA, J. HASEGAWA, M. ISHIDA, T. NAKAJIMA, Y. HONDA, O. KITAO, H. NAKAI, T. VREVEN, J. A. MONTGOMERY, JR., J. E. PERALTA, F. OGLIARO, M. BEARPARK, J. J. HEYD, E. BROTHERS, K. N. KUDIN, V. N. STAROVEROV, R. KOBAYASHI, J. NORMAND, K. RAGHAVACHARI, A. RENDELL, J. C. BURANT, S. S. IYENGAR, J. TOMASI, M. COSSI, N. REGA, J. M. MILLAM, M. KLENE, J. E. KNOX, J. B. CROSS, V. BAKKEN, C. ADAMO, J. JARAMILLO, R. GOMPERTS, R. E. STRATMANN, O. YAZYEV, A. J. AUSTIN, R. CAMMI, C. POMELLI, J. W. OCHTERSKI, R. L. MARTIN, K. MOROKUMA, V. G. ZAKRZEWSKI, G. A. VOTH, P. SALVADOR, J. J. DANNENBERG, S. DAPPRICH, A. D. DANIELS, Ö. FARKAS, J. B. FORESMAN, J. V. ORTIZ, J. CIOSLOWSKI, and D. J. FOX, Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT, 2009.
Patrocinadores





Apoio





Realização
