ISBN 978-85-85905-10-1
Área
Iniciação Científica
Autores
Silva, A.G. (UNINASSAU) ; Machado, C.M.B. (UFPE) ; Silva, R.P.G. (UFPE) ; Lima, N.B. (UFPE)
Resumo
Cálculos teóricos MP2/6-31++G (d, p) e B3LYP/6-31++G (d, p) foram realizados no sentido de obter energias de estabilidade e propriedades moleculares de complexos entre 3-aminoftalimida e HF. Os cálculos ab initio de orbitais moleculares e DFT revelaram que cinco diferentes complexos podem ser formados e que as geometrias otimizadas correspondem a pontos de mínimo de energia na curva de energia potencial. O processo de formação das ligações de hidrogênio ocasionaram mudanças nas propriedades da imida cíclica e do ácido linear monoprótico, sendo para este último os efeitos mais fortes.
Palavras chaves
imidas cíclicas; ligação de hidrogênio; ácido fluorídrico
Introdução
Cálculos ab initio de orbitais moleculares são frequentemente usados em simulações das propriedades moleculares e espectroscópicas de complexos de ligação de hidrogênio entre moléculas aceitadores de próton e moléculas doadoras de próton (LIMA et al, 2012). As imidas cíclicas podem são moléculas que tanto podem agir como aceitadoras de próton, uma vez que possuem grupos C=O quanto podem agir como doadoras de próton a partir do grupo N-H (LIMA et al, 2011). As imidas cíclicas são excelentes agentes biológicos e terapêuticos por exemplo, a ftalimida quando administrada em doses de 10 à 20 mg/Kg/dia em ratos, reduzem em poucos dias as taxas de colesterol e triglicerídeos destes. As atividades biológicas das imidas cíclicas estão diretamente relacionadas com o grupo imida cíclica, onde ao substituir alguns elementos deste grupo é possível potencializar os efeitos das atividades biológicas. Neste trabalho, nós realizamos cálculos ab initio de orbitais moleculares e DFT para a interpretação das propriedades estruturais, energéticas e vibracionais de complexos entre 3-amiftalimida, figura 1, e HF na proporção estequiométrica, imida cíclica: ácido 1:1.
Material e métodos
Realizamos, cálculos de orbitais moleculares usando a teoria do funcional de densidade (DFT) (BECKE, 1993) com o funcional B3LYP (GEERLINGS et al, 2003) com o conjunto de funções base 6-31++G(d,p) para otimização completa de geometria e cálculo dos modos vibracionais dos complexos. Todos os cálculos foram realizados utilizando o programa de química quântica computacional GAUSSIAN 2009 (FRISCH et al, 2009).
Resultado e discussão
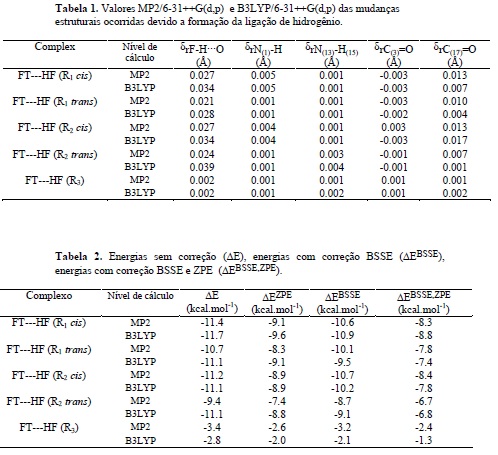
A tabela 1 apresenta as principais mudanças nas propriedades estruturais
observadas a partir dos cálculos computacionais. Em todos os casos que os
comprimentos da ligação H-F e da ligação N-H foram aumentados após a formação
dos complexos. Entretanto, o efeito foi mais intenso para a ligação do ácido
linear monoprótico. Provavelmente isto ocorreu porque a ligação de hidrogênio
ocorre entre o ácido HF e um dos grupos C=O da imida cíclica.
A tabela 2 apresenta os valores das energias das ligações de hidrogênio sem
correções (ΔE), com correção BSSE (∆EBSSE), energias com correção BSSE e ZPE
(∆EBSSE,ZPE). Os resultados mostram que preferencialmente a ligação de
hidrogênio é formada nas regiões 1 e 2 nas posições cis e trans em relação ao
grupo NH da imida cíclica. Isto ocorre porque a complexação na região 3 o ácido
HF é a molécula aceitadora de próton, e a imida cíclica age como a molécula
doadora de próton onde a interação ocorre no grupamento NH2, sendo este grupo
pouco eficiente na complexação.
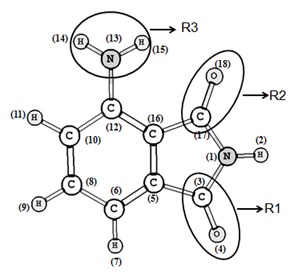
Estrutura do composto 3-aminoftalimida.
Conclusões
Os cálculos ab initio de orbitais moleculares e DFT revelaram que cinco diferentes complexos podem ser formados e que as geometrias otimizadas correspondem a pontos de mínimo de energia na curva de energia potencial. Preferencialmente, a complexação ocorre nos grupos C=O da imida cíclica. O processo de formação das ligações de hidrogênio ocasionaram mudanças nas propriedades da imida cíclica e do ácido linear monoprótico, sendo para este último os efeitos mais fortes.
Agradecimentos
Os autores agradecem ao CNPq, à FACEPE, ao PET/CAPES, à PROAES/UFPE e ao PRONEX/FACEPE.
Referências
Belarmino, M. K. D. ; LIMA, N. B. ; RAMOS, M. N. . Hydrogen bonds between acetylene and formic acid: an ab initio study. International Journal of Quantum Chemistry, v. 112, p. 3246-3251, 2012.
N.B. de Lima, V.H. Rusu, M.N. Ramos, Hydrogen bonds between phthalimide and hydrogen fluoride: A theoretical study, Int. J. Quantum Chem. 111 1387–1394, 2011.
A.D. BECKE, Density‐functional thermochemistry. III. The role of exact Exchange. The Journal of Chemical Physics, Vol. 98, P. 5648, 1993.
P. GEERLINGS, F. DE PROFT, W. LANGENAEKER, Conceptual Density Functional Theory. Chemical Reviews. Vol. 103, P. 1793–1874, 2003.
M. J. FRISCH, G. W. TRUCKS, H. B. SCHLEGEL, G. E. SCUSERIA, M. A. ROBB, J. R. CHEESEMAN, G. SCALMANI, V. BARONE, B. MENNUCCI, G. A. PETERSSON, H. NAKATSUJI, M. CARICATO, X. LI, H. P. HRATCHIAN, A. F. IZMAYLOV, J. BLOINO, G. ZHENG, J. L. SONNENBERG, M. HADA, M. EHARA, K. TOYOTA, R. FUKUDA, J. HASEGAWA, M. ISHIDA, T. NAKAJIMA, Y. HONDA, O. KITAO, H. NAKAI, T. VREVEN, J. A. MONTGOMERY, JR., J. E. PERALTA, F. OGLIARO, M. BEARPARK, J. J. HEYD, E. BROTHERS, K. N. KUDIN, V. N. STAROVEROV, R. KOBAYASHI, J. NORMAND, K. RAGHAVACHARI, A. RENDELL, J. C. BURANT, S. S. IYENGAR, J. TOMASI, M. COSSI, N. REGA, J. M. MILLAM, M. KLENE, J. E. KNOX, J. B. CROSS, V. BAKKEN, C. ADAMO, J. JARAMILLO, R. GOMPERTS, R. E. STRATMANN, O. YAZYEV, A. J. AUSTIN, R. CAMMI, C. POMELLI, J. W. OCHTERSKI, R. L. MARTIN, K. MOROKUMA, V. G. ZAKRZEWSKI, G. A. VOTH, P. SALVADOR, J. J. DANNENBERG, S. DAPPRICH, A. D. DANIELS, Ö. FARKAS, J. B. FORESMAN, J. V. ORTIZ, J. CIOSLOWSKI, and D. J. FOX, Gaussian 09, Revision D.01. Gaussian, Inc., Wallingford CT, 2009.
Patrocinadores





Apoio





Realização
