ISBN 978-85-85905-10-1
Área
Físico-Química
Autores
Costa, A. (UFMA) ; Gomes Varela Júnior, J.J. (UFMA) ; Pereira Silva, A.L. (UFMA) ; Matos Mourão Neto, I. (UFMA)
Resumo
No presente trabalho apresentamos um estudo das propriedades estruturais e eletrônicas da Porfirina de Manganês e Octametilporfirina de Manganês, onde foram empregados cálculos (DFT), utilizando o funcional híbrido B3LYP, com a utilização do programa GAUSSIAN 09. As funções de bases LANL2TZ(f) foram usadas para átomo de Mn, e 6-311G(d,p) usadas para os átomos de C, N e H. O Método B3LYP/L LANL2TZ(f),6-311G(d,p) descreveu o comportamento dos complexos de Mn (II) e os parâmetros geométricos calculados, estão de acordo com os dados experimentais da literatura. Além disso, ambas as moléculas apresentaram multiplicidade de spin sexteto como mais estáveis e gap 2,118 e 2,132 eV para os elétrons α e 2,886 e 2,769 eV para os elétrons β dos complexos MnP e MnOMP, respectivamente.
Palavras chaves
Energia relativa; Metaloporfirinas; DFT
Introdução
Complexos de metais de transição envolvendo ligantes macrocíclicos se destacam por representarem sistemas cineticamente inertes, estáveis do ponto de vista termodinâmico por apresentarem elevada seletividade (ELIAS, H 1999). Sendo assim, estudos envolvendo macrociclos de metais de transição, particularmente, metaloftalocianinas e metaloporfirinas, para a reação redução de oxigênio (RRO) em soluções aquosas têm sido bastante realizados, pois estes compostos pertencem a uma classe que fornecem oportunidades singulares para se examinar em detalhes os fatores envolvidos na ativação e posterior redução da molécula de oxigênio (MASA J. et al, 2012) . Desta forma, o presente trabalho apresenta um estudo introdutório das propriedades estruturais dos complexos MnP (Porfirina de Manganês) e MnOMP (Porfirina de Manganês Substituída - Octametilado).
Material e métodos
No presente trabalho, foi escolhido o metal Mn como centro metálico para os macrocíclicos MnP e MnOMP. Os cálculos de otimização da geometria e frequências vibracionais das estruturas foram realizadas no programa GAUSSIAN 09, utilizando o funcional B3LYP, com as funções de base LANL2TZ(f) para o (Mn) e 6-311G(d,p) para (H, C, e N). A verificação das possíveis multiplicidades de spin para os complexos foram testadas, com intuito de determinar o estado mais estável.
Resultado e discussão
Na tabela 1 observou-se que o estado fundamental para os complexos MnP e MnOMP
foram o sexteto, como já era esperado baseado em estudos teóricos (O.P.CHARKIN
et al, 2008). O estado dupleto e quarteto para a molécula MnP apresentaram
diferenças energéticas, em relação ao estado sexteto, de 179,93 kJ mol-1 e
27,34 kJ mol-1 enquanto que para a molécula MnOMP as diferenças energéticas
foram da ordem de 230,42 kJ mol-1 e 27,48 kJ mol-1. Outro fato importante que
não observamos foram distorções com relação à planaridade do anel porfiranato,
sendo estes complexos de simetria D4h, corroborando com os resultados
experimentais para estruturas similares determinadas por difração de raios-X
(B.GONZALEZ et al, 1975)que apresentou uma distância Mn–N de 2,082 Å.
A energia do gap (diferença de energia do mais alto orbital ocupado HOMO e do
mais baixo orbital desocupado LUMO) é um importante índice de estabilidade e/ou
reatividade da molécula. Sendo assim, com o intuito de mostrar o gap de energia
nos orbitais moleculares (HOMO-LUMO), foram construídos gráficos com os cinco
últimos orbitais moleculares de fronteira ocupados (HOMO) e os cinco primeiros
orbitais moleculares de fronteira desocupado (LUMO), dos elétrons alfa (α) e
beta (β), para os complexos em estudados.
Nesse contexto, pode-se observar na Figura 2 o valor do gap 2,118 e 2,132 eV
para os elétrons α e 2,886 e 2,769 eV para os elétrons β dos complexos MnP e
MnOMP. Como pode ser analisado o valor do gap para os complexos MnP e MnOMP foi
aproximadamente igual nos elétrons α e menor se comparado aos dos elétrons β
dessa forma os compostos para os elétrons α apresentam uma maior reatividade
para a redução de redução do oxigênio .
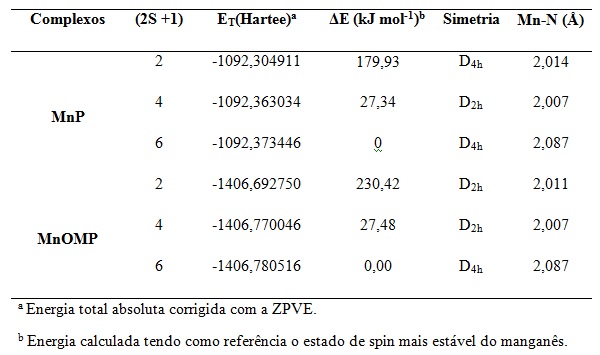
Multiplicidades (2S+1), energia total (ET) em Hartree e energia relativa (ΔE) em kJ mol-1, comparada para os complexos MnN4.
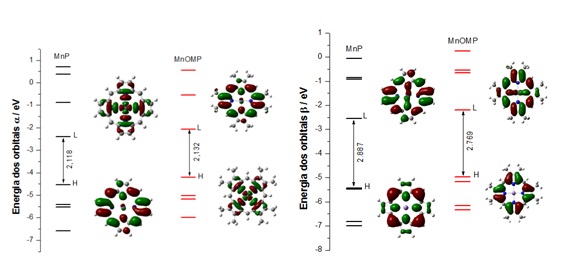
Diagrama energético para os elétrons alfa (α) e beta (β) dos cinco primeiros orbitais para os compostos MnP, MnOMP.
Conclusões
A metodologia empregada nos cálculos, B3LYP/ LANL2TZ(f)/6-311G(d,p) em nível de DFT, descreve satisfatoriamente o comportamento dos compostos Porfirina de Manganês e Octametilporfirina de Manganês. De maneira que os parâmetros calculados estão de acordo com os resultados obtidos experimentalmente e disponíveis na literatura apresentando multiplicidade de spin sexteto mais estáveis para MnP e MnOMP. Neste sentido observamos também o menor gap de energia HOMO-LUMO, para os complexos dos orbitais α dessa forma os complexos apresentaram uma maior atividade para a ligação com O2.
Agradecimentos
A Universidade Federal do Maranhão, CNPq e a FAPEMA.
Referências
B.GONZALEZ.;J.KOUBA.S.YEE.C.A.REED.J.F.KINER.;W.R.SCHEIDT,J.Am.Chem.Soc.97.32 7 (1975).
ELIAS, H.; Coordination Chemistry Reviews; 187, 37-73, 1999.
O.P.CHARKIN.;A.V.MARKAROV.;N.M.KLIMENKO.; Theoretical Study of First-Row Transition Metal Porphyrins and Their Carbonyl Complexes. Theoretical Inorganic Chemistry, 2008.Vol.53,No.5,pp.716-728.
MASA.J.; OZOEMENA.K.;SCHUHMANN W.; ZAGAL.J.Oxygen reduction reaction using N4-metallomacrocyclic catalysts: Fundamentals on Rational Catalyst Design. Journal. Porphyrins Phthalocianines 2012; 16: 761-784.
Patrocinadores





Apoio





Realização
