ISBN 978-85-85905-10-1
Área
Físico-Química
Autores
Lima, E.M.A. (UFRN) ; Vieira, D.S. (UFRN)
Resumo
As xilanases hidrolisam a cadeia glicosídica da xilana contida na superfície da polpa, facilitando a extração da lignina [1]. O processo de inibição de enzimas xilanases GH11 homólogas (NpXyn e Xync) através do acoplamento proteína-proteína, foi investigado por simulação molecular. A xilanase NpXyn é um caso incomum de insensibilidade à proteína inibidora XIP-I [2], e portanto, um ótimo modelo para identificar as características estruturais responsáveis pela resistência à inibição. O estudo comparativo de xilanases homólogas com diferentes respostas à inibição é fundamental para o entendimento e caracterização do sítio ativo dessas enzimas, visto que esforços têm sido feitos para otimizar a estabilidade e atividade dessas enzimas para aplicação industrial.
Palavras chaves
Simulação Molecular; Xilanase; Inibição
Introdução
Proteinas são macromoléculas flexiveis cuja movimentação está é determinante para o entendimento da sua atividade biológica. Os diferentes tipos de movimentos que a proteína exibe são responsáveis pela ligação de um subtrato, adaptação para diferentes condições, agregações, ajustes conformacionais em efeitos alostéricos e etc. Estas movimentações são chamadas de movimentos funcionais. Os movimentos funcionais envolvem complexas correlações entre as movimentações atômicas, que são determinadas pelas interações intramoleculares. O desafio é derivar tais movimentações das interações entre os atomos da proteina, com o intuito de identificar a sua funcionalidade e reduzir a dinâmica complexa da proteina para seus graus enssenciais de liberdade [3,4]. Xilanases GH11 são enzimas produzidas por vários organismos como bactérias, fungos, algas, protozoários e artrópodes e são capazes de hidrolisar as ligações 1,4-β de cadeia principal da xilana, principal componente hemicelulósico da parede celular vegetal [5]. Xilanases têm sido utilizadas no processo de branqueamento do papel e na produção de bioetanol. Vários tipos distintos de inibidores de xilanases GH11 foram descobertos a partir de extratos de proteína da farinha de trigo, o mais efetivo para as xilanases GH11 é a proteína XIP-1. Foi demonstado que a XIP-1 não funciona para uma xilanase GH11 (NpXyn) produzida pelo fungo Neocallimastix patriciarum, e os fatores estruturais/evolutivos responsáveis ainda não foram estabelecidos. Dentro desse contexto, o presente trabalho apresenta um estudo por simulações de dinâmica molecular de dois complexos protéicos formados pelas xilanase GH11 homólogas NpXyn11 e Xync, a última, produzida pela bactéria Penicillium funicolosum, sofre inibição pela XIP-1.
Material e métodos
Neste trabalho investiga-se por simulações de dinâmica molecular, o processo de inibição de enzimas xilanases GH11 através do acoplamento proteína-proteína. As redes de interações intermoleculares proteína-proteína serão investigadas com o intuito de identificar os principais resíduos de aminoácidos responsáveis pela estabilização da interface protéica. O estudo irá nos possibilitar o entendimento da base estrutural do processo inibitório desta família de enzimas, possibilitando assim, uma completa caracterização estrutural/energética dos sítios ativos de enzimas xilanases homólogas. Os sistemas foram simulados usando o programa de dinâmica molecular GROMACS 4.5.5 [6] e modelados pelo campo de força GROMOS96 (43A1) [7], sendo que o modelo SPC [8] foi usado para descrever as moléculas de água. Os sistemas foram simulados no ensemble NVT, a 298K, usando o algoritmo de Berendsen para controlar a temperatura. O algoritmo LINCS foi usado para restringir a distância de ligação com átomos de hidrogênio, e o SETTLE para restrição da geometria da molécula de água. O algoritmo Leap-Frog foi usado para integrar a equação de movimento com tempo de integração de 2.0fs. As interações de longo alcance foram tratadas usando PME com raio de corte de 1,2 nm. As estruturas iniciais dos complexos foram obtidas do banco de dados de proteínas RCSB (www.rcsb.org/pdb), a NpXyn com o código de 2C1F e a proteína inibidora XIP-I com o código 1OMO. Já a enzima XYNC está registrada sob código 1UKR. E o complexo XYNC-XIP-I, código 1TE1.
Resultado e discussão
A figura 1 apresenta a estrutura tridimensional do complexo NpXyn-XIP-I destacando as regiões da xilanase usando a conhecida analogia com a mão direita para denominá-las. A estabilidade estrutural da NpXyn complexada com a XIP-I, foi alcançada em 35ns e a XIP-I em 55ns, com o raiz quadrada do desvio quadrático médio (RMSD) apresentando um valor em torno de 0,21nm e 0,31nm, respectivamente. A estabilidade estrutural da Xync complexada com a XIP-I, foi alcançada em 90ns e a XIP-I em 35ns, com o RMSD apresentando um valor em torno de 0,22nm e 0,30nm, respectivamente. A xilanase NpXyn11.
O potencial de interação médio para os acoplamentos Npxyn+XIP-I e Xync+XIP-I são -1058,57 kJ/mol e -784,17 KJ/mol, respectivamente. A flexibilidade foi analisada pela raiz quadrática da flutuação quadrática média (RMSF) usando os átomos de carbonos alfa como base para os cálculos. A figura 2 apresenta o RMSF por resíduo para ambas xilanases. O RMSF mostra claramente diferentes flexibilidades em diferentes regiões de contato direto das xilanases NpXyn e Xync com o XIP-1. Os diferentes perfis de flexibilidade apresentado por essas xilanases em contato com o mesmo inibidor é um potencial indicativo que o processo de inibição está sendo governado, essencialmente, pela dinâmica e encaixe das partes envolvidas.
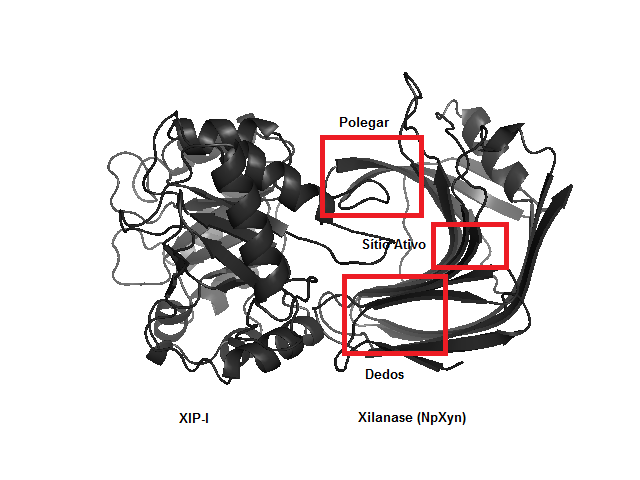
Estrutura tridimensional do complexo NpXyn/XIP-1 destacando as principais regiões, com analogia à mão direita.
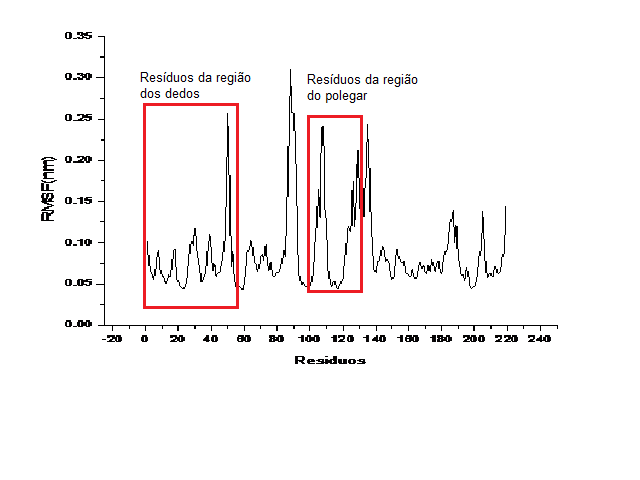
RMSF para as xilanases NpXyn). As regiões descritas na figura 1 estão indicadas aqui.
Conclusões
O trabalho apresentado trata do acoplamento proteína-proteína por dinâmica molecular, indicando que, nesse caso, a inibição parece estar melhor relacionada com o “encaixe” que envolve determinadas regiões da xilanase (regiões dedo e polegar) com o inibidor XIP-1, do que diretamente relacionada com a energia de interação média total entre eles. As ligações de hidrogênio estão sendo mapeadas, assim como a aplicação de estudos computacionais utilizando alanine scanning estão sendo realizados para identificar os hot-spots na interface protéica, para então, avaliar as mais importantes interações.
Agradecimentos
Pró-reitoria de pesquisa da Universidade Federal do Rio Grande do Norte – PROPESQ/UFRN
Referências
[1]T. K. Kirk, T. W. Jeffries, Enzimes for Pulp and Paper Processing, American Chemical Society, (1996).
[2] Payan, F., Leone, P., Porciero, S., Furniss, C., Tahir, T.,Williamson, G. et al. (2004). The dual nature of the wheat xylanase protein inhibitor XIP-I: structural basis for the inhibition of family 10 and family 11 xylanases. J. Biol. Chem. 279, 36029–36037.
[3] A. Amadei, A. B. M. Linsen, B. L. de Groot, D. M. F. van Aalten, J. H. C. Berendsen, J.Struc. Dyn, 13,615, (1996) [4] A. Amadei, A. B. M. Linsen, J. H. C. Berendsen, Essential dynamics of proteins. Proteins Struc. Funct. Genet. 17, 412, (1993).
[5] Brett, C. T. & Waldren, K. (1996). Physiology and biochemistry of plant cell walls. In Topics in Plant Functional Biology (Black, M. & Charlewood, B., eds), vol. 1, Chapman and Hall, London
[6] E. Lindahl, B. Hess, D. van der Spoel. (2010)
[7] W. F. van Gunsteren, S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott, I. G. Tironi, Biomolecular Simulation: The GROMOS96 Manual and User Guide, Biomos, Groningen, (1996)
[8] H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, J. Hermans, Interactions models for water in relation to preotein hydration. In:Intermolecular Force. Pullman, B. ed Reidel Publishing Company Dordrecht, 331,(1981)
Patrocinadores





Apoio





Realização
