ISBN 978-85-85905-15-6
Área
Química Inorgânica
Autores
Sousa, K.M.D. (UFMT) ; Chagas, M.A.S. (UNB) ; Santos, J.M.F.D. (UFMT) ; Santos, W.B. (UFMT)
Resumo
A utilização de técnicas computacionais para estudo e comparação teórica através de aproximações matemáticas foi objetivo deste trabalho. Para comparação teórica do complexo [RuCl3(H2O)2(Gly)] em relação aos dados experimentais, foram realizados cálculos de otimização da geometria e frequências vibracionais harmônicas para as três possíveis geometrias provenientes do complexo, de forma a determinar dentre elas, a mais estável energeticamente e as frequências de maior precisão com o experimental.
Palavras chaves
DFT; Frequências vibracionais; Complexo de Rutênio III
Introdução
A utilização de softwares computacionais baseados em química quântica tem crescido bastante nas últimas décadas entre parte da comunidade científica, pois tem mostrado resultados expressivos no tratamento de vários sistemas desde os mais simples até os mais complexos, como organometálicos, fármacos, polímeros, biomoléculas, entre outros, assim como suas interações em sistemas biológicos como forma de auxílio na interpretação de dados experimentais para melhor compreensão dos resultados (Ramachandran, Deepa et al. 2008). A síntese e caracterização do complexo [RuCl3(H2O)2(Gly)] apresentado no trabalho de (Chagas 2015), indica que a molécula deve ser estudada mais afundo, pois ela apresentou coordenação com guanina, base púrica do DNA, e interação com algumas células tumorais. A molécula também mostrou atividade leishmanicida apresentada por (SALAMA 2013) em concentrações elevadas com teste em Camundongos isogênicos da linhagem BALB/c. Devido ao potencial biológico apresentado pelo composto [RuCl3(H2O)2(Gly)] é necessário estudos mais aprofundados e principalmente a determinação de sua estrutura geométrica, no intuito de conduzir à modificações estruturais como forma de aumentar seu potencial biológico. E como obter a geometria de uma molécula? É possível através da análise por difração de raio X, porém é uma técnica que requer um trabalho experimental mais complexo para a obtenção do monocristal necessário para análise. Neste trabalho apresentamos o recurso computacional como alternativa para elucidação da geometria da molécula em estudo, uma vez que tem se mostrado promissor tanto em estudos de comparação quanto de investigação da estrutura e de interações atômicas e moleculares.
Material e métodos
Todos os cálculos foram realizados utilizando o pacote de programas Gaussian 09 (Frisch, Trucks et al. 2009). As geometrias foram otimizadas pelo método DFT (Teoria do Funcional da Densidade) e o funcional corrigido Método de Coulomb Atenuado do híbrido funcional B3LYP (CAM-B3LYP)(Yanai, Tew et al. 2004). As bases utilizadas foram 6-31G(d) para todos os átomos com exceção do Rutênio que foi tratado com o conjunto de base SDD (Stuttgart/Dresden duplo-ζ) com ECP (potencial efetivo do núcleo), no qual trata os elétrons mais internos do átomo de Rutênio (Andrae, Häußermann et al. 1990). As frequências vibracionais harmônicas foram calculadas com segundas derivadas analíticas, sem correções anarmônicas. Todos os cálculos foram realizados na Universidade Federal do Mato Grosso, Laboratório de Estudos de Materiais.
Resultado e discussão
Através dos dados de otimização foi possível obter o mínimo de energia
possível para cada estrutura geométrica da molécula [RuCl3(H2O)2(Gly)],
dentre elas temos: cis-fac-diaquotriscloroglicinatorutênio(III) (molécula I)
com energia igual à -1912.6881691 Hartree, cis-mer-
diaquotriscloroglicinatorutênio(III) (molécula II) com energia igual à
-1912.6726627 Hartree e trans-mer-diaquotriscloroglicinatorutênio(III)
(molécula III) com energia igual à -1912.6811518 Hartree; como essas
moléculas tem energias muito próximas, isto é um indicativo da existência
das três, porém em função da energia mais baixa, a molécula I teoricamente
se mostrou mais estável do que as outras duas. Comparando os dados teóricos
e experimentais relacionados à espectroscopia no infravermelho próximo
(tabela 1), observamos que a molécula 1 é a que mais se aproxima dos dados
experimentais, principalmente em relação aos grupos (COO-) estiramento
simétrico, (NH3+) dobramentos e deformações, (CCN) estiramento e (CH2)
estiramento e deformações. Os dados equivalentes aos picos vibracionais
(OH2), não foram descritos experimentalmente, porque não era o foco inicial
do trabalho de (Chagas 2015), ficando restrita a coordenação da glicina com
o metal, porém é possível observar em seu trabalho um pico largo de fraca
intensidade próxima a região de 3500 cm-1. Alguns valores vibracionais
calculados se mostram altos em função da não inclusão do efeito de
anarmonicidade (Nakamoto 2008) que proveria resultados mais próximos do
experimental. Algumas frequências correspondentes aos modos vibracionais de
estiramento e deformação para o grupo (CH2) não foram apresentados na tabela
devido as suas baixas intensidades. Para considerações de energia 1 Hartree
equivale à 627.5095 kcal-mol-1 ou 27.2114 eV (Mohr, Taylor et al. 2008).
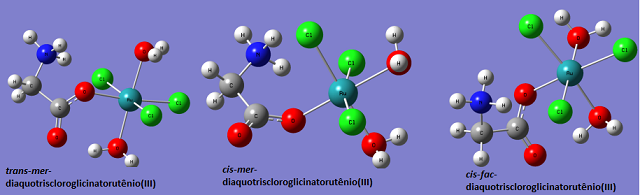
Imagem das três moléculas obtido pelo programa GaussView 5.0
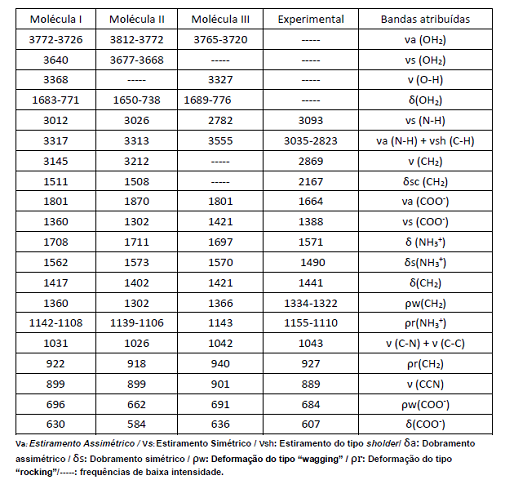
Frequências calculadas por DFT dos isômeros [RuCl3(H2O)2(Gly)], e FTIRmed (4000-600 cm-1) com atribuição aproximada de bandas.
Conclusões
Os cálculos realizados mostraram em acordo com os dados experimentais, indicando que a geometria proposta na molécula 1, cis-fac- diaquotriscloroglicinatorutênio(III), como sendo a molécula sintetizada por (Chagas 2015), mesmo com a ausência do efeito de anarmonicidade nos cálculos de frequência vibracional harmônica, alguns valores tiveram boa aproximação. Esta técnica mostrou-se eficaz para estudos comparativos e para trabalhos futuros do âmbito investigativo, uma ferramenta que possa auxiliar e complementar pesquisas de caráter elucidativo.
Agradecimentos
Trabalho apoiado pela CAPES.
Referências
Andrae, D., U. Häußermann, M. Dolg, H. Stoll and H. Preuß (1990). "Energy-adjustedab initio pseudopotentials for the second and third row transition elements." Theoretica chimica acta77(2): 123-141.
Chagas, M. A. S. (2015). "Síntese e caracterização de compostos de Ru3+ com ligantes: Glicina e Guanina e avaliação do efeito antitumoral." Tese de Mestrado, Universidade Federal do Mato Grosso,Cuiabá-Mato Grosso, 2015.
Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox (2009). Gaussian 09. Wallingford, CT, USA, Gaussian, Inc.
Mohr, P. J., B. N. Taylor and D. B. Newell (2008). "CODATA recommended values of the fundamental physical constants: 2006*." Reviews of Modern Physics80(2): 633-730.
Nakamoto, K. (2008). Infrared and Raman Spectra of Inorganic and Coordination Compounds, Theory and Applications in Inorganic Chemistry, Wiley.
Ramachandran, K. I., G. Deepa and K. Namboori (2008). "Computational Chemistry and Molecular Modeling " Springer-Verlag Berlin Heidelberg1: 398.
SALAMA, I. C. C. D. A. (2013). "ANÁLISE BIOLÓGICA DOS COMPOSTOS RUTÊNIO, LUPEOL E BOLDINA NO MODELO EXPERIMENTAL DAS LEISHMANIOSES." Tese de Mestrado, Universidade Federal do Mato Grosso-Campus Universitário do Araguaia, 2013.
Yanai, T., D. P. Tew and N. C. Handy (2004). "A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP)." Chemical Physics Letters393(1–3): 51-57.
Patrocinadores







Apoio









Realização

