ISBN 978-85-85905-15-6
Área
Físico-Química
Autores
Pinheiro, J.C. (UFPA) ; Santos, M.A.B. (UEPA) ; Consolação, W.A. (UFPA) ; Brito, H.G. (UFPA) ; Alves, S.S.S. (IFPA) ; Maciel, A.A. (SEDUC)
Resumo
Este trabalho utilizou os métodos da química computacional e quimiometria para estudar derivados do composto espiro e diespiro-1,2,4-trioxolano com atividade antimalárica contra a cepa K1 do Plasmodium falciparum. As estruturas moleculares foram otimizadas a um mínimo global com o método B3LYP/6-31G*. Mapas de potencial eletrostático molecular auxiliaram na identificação de características relacionadas à atividade biológica destes compostos. Procedimento de docking molecular foi realizados para estudar os complexos formados entre o ligante e receptor (heme).
Palavras chaves
Química computacional; mapas MEP; complexos
Introdução
A malária é uma doença infecciosa causada pelo parasita protozoário Plasmodium, sendo endêmica em regiões tropicais africanas, asiáticas e sulamericanas. Esta doença provocou aproximadamente 584 mil mortes de malária em todo o mundo durante o ano de 2013. Todos os anos são registrados aproximadamente mais de 198 milhões de casos, sendo a grande maioria na África Sub-Saariana. As crianças de até 5 anos de idade são as mais afetadas, e 78% do total chega a óbito. Cerca de 40% da população mundial, distribuídos em 107 países, estão nas áreas consideradas de risco de contrair a doença, que está controlada em áreas de clima temperado, mas ainda ativa nas regiões tropicais e subtropicais (WHO, 2013).
Material e métodos
Este trabalho é um estudo computacional sobre moléculas derivadas da adamantano que possuem atividade antimalárica contra a cepa K1 do P. falciparum. Estas são derivados do espiro e diespiro-1,2,4-trioxano com atividades antimaláricas foram selecionadas da literatura (YUXIANG et al., 2006). Essas estruturas foram desenhadas e submetidas a cálculos de estrutura eletrônica utilizando o método semi-empírico AM1 e em seguida as geometrias obtidas foram otimizadas a um mínimo global pelo método B3LYP em conjunto com a base de valência separada 6-31G*. A partir das estruturas otimizadas, os mapas de potencial eletrostático (MEP) e as cargas ESP foram extraídas dos cálculos de estrutura eletrônica single point. Os mapas MEP foram criados com o auxílio do programa Molekel 5.4 e as propriedades utilizadas no modelo QSAR foram calculadas pelo software MarvinChem 5.3.1 (CHEMAXON, 2010) e pelo ambiente virtual E-Dragon 1.0 (TETKO et al., 2005). O estudo do docking molecular foi realizado pelo programa AutoDock4 (MORRIS et al., 2009). As análises estatísticas foram executas pelo programa Pirouette 3.10 (INFOMETRIX, 2001).
Resultado e discussão
As moléculas dos compostos derivados do espiro e diespiro-1,2,4-trioxano com atividades antimaláricas foram selecionadas da literatura (YUXIANG et al., 2006).Essas estruturas foram desenhadas e submetidas a cálculos de estrutura eletrônica utilizando o método semi-empírico AM1 e em seguida as geometrias obtidas foram otimizadas a um mínimo global pelo método B3LYP em conjunto com a base de valência separada 6-31G*. A partir das estruturas otimizadas, os mapas de potencial eletrostático (MEP).Os mapas MEP, foram criados com o auxílio do programa Molekel 5.4 e as propriedades utilizadas no modelo QSAR foram calculadas pelo software MarvinChem 5.3.1 (CHEMAXON, 2010) e docking molecular, foi realizado pelo programa AutoDock4 (MORRIS et al., 2009). As análises estatísticas foram executas pelo programa Pirouette 3.10 (INFOMETRIX, 2001).
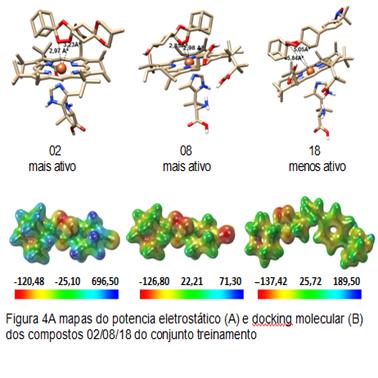
mapas de potencial eletrostatico molecular e docking molecular
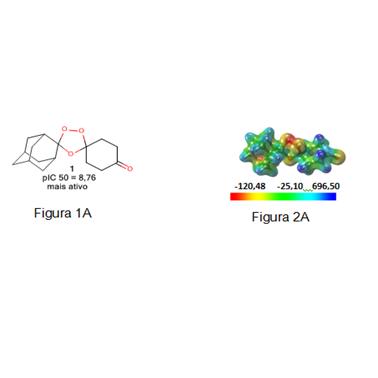
figuras referentes a base não otimizada e o mapa de potencial eletrostático molecular
Conclusões
Este trabalho baseia-se no estudo dos parâmetros físico-químicos que são determinantes na ação de antimaláricos peroxídicos, derivados do composto espiro e diespiro-1,2,4-trioxano.O cálculo das cargas nos oxigênios mostrou uma forte dependência da atividade com essa propriedade, indicando maiores afinidades eletrônicas e isto é essencial no processo de transferência de elétrons do Fe2+ para os oxigênios peroxídicos.O estudo de docking molecular dos antimaláricos estudados confirma a regiosseletividade de ataque do heme ao oxigênio peroxídico estericamente menos impedido. Essa regiosseletividade já poderia ser prevista pela carga mais negativa localizada no O2 da ligação peróxido, já que sabemos que as regiões de maiores densidades eletrônicas estão localizados na ligação peróxido.
Agradecimentos
Ao Laboratório de Química Teórica e Computacional da Universidade Federal do Pará (LQTC)
Referências
ABE, M. et al. 18O-Tracer Studies of Fe(II)-Induced Decomposition of 1,2,4-Trioxolanes (Ozonides) Derived from Cyclopentenes and Indenes. Inner-Sphere Electron Transfer Reduction of the Peroxide Linkage. Journal of the American Chemical Society. v. 121(28), p. 6556-6562, 1999.
ANDERSSON, C. D. et al. A Multivariate Approach to Investigate Docking Parameters Effects on Docking Performance. Journal of Chemical Information and Modeling. v. 47(4), p. 1673–1687, 2007.
ARAÚJO, J. Q. et al. Interaction between artemisinin and heme. A density functional theory study of structures and interaction energy, Bioorganic & Medicinal Chemistry v. 16, p. 5021-5029, 2008.
Patrocinadores







Apoio









Realização

