ISBN 978-85-85905-15-6
Área
Físico-Química
Autores
Vaz, E.C. (UNIVERSIDADE ESTADUAL DE GOIÁS) ; Camargo, A.J. (UNIVERSIDADE ESTADUAL DE GOIÁS) ; Oliveira, S.S. (UNIVERSIDADE ESTADUAL DE GOIÁS)
Resumo
O nimesulida é um medicamento, que possui fórmula molecular C13H12N2O5S, cujo nome 4-nitro-2-fenoxi-metanosulfonanilida serviu de base para o nome genérico do fármaco. É da classe dos anti-inflamatórios não esteroides (AINEs), que atua através da inibição da enzima ciclooxigenase (COX). Desta forma, o nimesulida combate os processos inflamatórios, as dores e a febre. Realizou-se estudos teóricos para verificar o comportamento molecular, as propriedades geométricas e eletrônicas do nimesulida no vácuo, por meio da dinâmica molecular de Car-Parrinello e pela Teoria do Funcional de Densidade (DFT). Realizou-se os cálculos com o auxílio do programa Quantum Espresso para Car-Parrinello e do Gaussian 09 para DFT.
Palavras chaves
Nimesulida; Car-Parrinello; Dinâmica molecular
Introdução
O nimesulida é um fármaco da classe dos anti-inflamatórios não-esteroides (AINEs) e atua inibindo a biossíntese de prostaglandinas, através de um bloqueio competitivo da enzima ciclooxigenase (COX) e também diminuindo a formação de radicais livres em nível da cascata do ácido araquidônico, impedindo a formação de eicosanoides, combatendo, assim, os processos inflamatórios, dores e febre [1,2]. Realizamos a dinâmica de Car-Parrinello no vácuo objetivando analisar as propriedades geométricas da molécula. Para comparação, também realizamos cálculos de Teoria do Funcional de Densidade (DFT), e analisamos a estrutura eletrônica. Comparamos nossos resultados com dados experimentais obtidos por difração de raios-X. Modificações em sua estrutura molecular foram listadas na literatura provando um maior efeito anti-inflamatório do nimesulida [3-5]. Tais modificações juntamente dos resultados obtidos serão utilizados para estudos de uma dinâmica em sistema aquoso focando melhorias na sua farmacodinâmica. Para estudo do nimesulida, utilizou-se a Dinâmica Molecular de Car-Parrinello (DMCP), pois a mesma é uma combinação das vantagens de duas outras dinâmicas; de Ehrenfest (DME); e de Born-Oppenheimer (DMBO). Sendo assim, a DMCP é capaz de: 1) Calcular propriedades do estado fundamental de sistemas em nível de cálculo de estrutura eletrônica; 2) Realizar simulações utilizando mecânica clássica para o movimento iônico e a aproximação de Born-Oppenheimer para separar coordenadas nuclear e eletrônica.
Material e métodos
Na DMCP é realizado simultâneamente a descrição clássica do movimento dos núcleos junto à resolução da equação de Schrödinger para os elétrons, e para conectar esses dois termos é utilizada a lagrangeana estendida de Car-Parrinello (Figura 2C). Na equação estão presentes dois termos de energia cinética, o funcional de energia de Kohn-Sham (energia potencial) e uma restrição de ortonormalidade. Para os cálculos, a molécula primeiramente foi otimizada geometricamente e minimizada sua função de onda com o auxílio do algoritmo Steepest Descent, e a molécula estabilizada foi utilizada como input para os cálculos em todos os métodos. Para Car-Parrinello a molécula foi disposta em uma caixa cúbica de 18Å de diâmetro e os cálculos foram realizados a uma temperatura de 300K com passo de integração de 5,0 a.t.u. e massa fictícia de 400 a.u., para os cálculos utilizou-se o programa Quantum Espresso. Para o método de Teoria do Funcional de Densidade (DFT), os cálculos com base 6-31+g(d,p) foram realizados com o auxílio do programa Gaussian 09. Os cálculos foram realizados em um sistema de cluster computacional pertencente à Universidade Estadual de Goiás.
Resultado e discussão
Os resultados da dinâmica mostraram-se positivos, confirmando que a molécula
possui comportamento estável, como pode ser observado na forma molecular do
nimesulida obtida através dos cálculos na figura 1A. Os resultados também
mostraram que a molécula manteve a separação energia entre os subníveis
eletrônico e iônico durante toda a simulação, como pode ser observado na
figura 1B. Por meio de DFT pode-se observar as propriedades eletrônicas do
nimesulida, tais como mapa de potencial eletrostático (MEP) e orbitais HOMO-
LUMO (1C, 1D). As comparações dos parâmetros geométricos, tais como
comprimento de ligação e ângulos de ligação, entre os métodos teóricos e os
resultados experimentais estão expressos nas figuras 2A e 2B. Ao observar a
figura 2A, verificamos que havia um grupo de valores que mais se distanciava
da reta em relação aos outros (pontos destacados no gráfico). Ao analisarmos
esses valores, verificamos que se trata de uma possível repulsão eletrônica
H-H, entre H(8)-H(12), H(9)-H(10)
e H(2)-H(7). Os demais valores apresentaram um
aceitável desvio padrão da reta. Já ao observarmos a figura 2B, verificamos
que os valores de ângulo de ligação estavam bastante dispersos, mas dois
grupos se destacavam mais, em relação aos outros, da reta (pontos destacados
no gráfico). Ao analisarmos os valores, percebemos que se trata do efeito de
eletropositividade na angulação da molécula, uma vez que os pontos que
apresentaram alto desvio padrão correspondem a angulação entre S(33)
-N(27)-H(9) e S(33)-N(27)-
C[sub(17) (como pode ser observado com o auxílio do mapa de potencial
eletrostático).
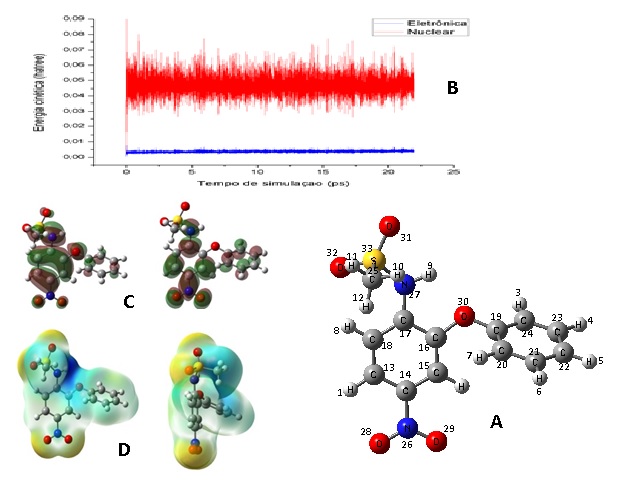
Sub-divida em: Figura 1A; 1B; 1C; 1D, para utilização como referência do resumo
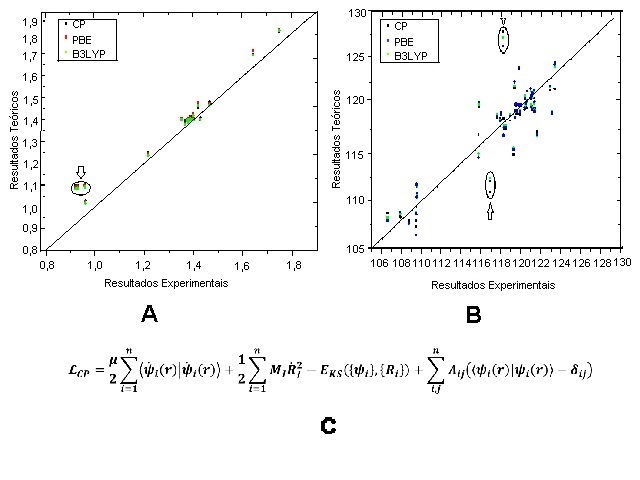
Sub-dividida em: 2A, 2B e 2C para utilização como referência no artigo.
Conclusões
Por meio dos resultados pode-se observar as regiões as quais o solvente provavelmente irá interagir. A probabilidade de interação é maior devido à presença de uma região eletropositiva na molécula e do posicionamento do orbital LUMO. Tal região tem maior facilidade em liberar próton, facilitando também a interação de um solvente com a molécula. Pode-se também comparar e verificar qual dos métodos possui maior acurácia quando comparado aos resultados experimentais a nível de parâmetros geométricos, sendo ele o método de Teoria do Funcional de Densidade B3LYP.
Agradecimentos
Agradeço a Universidade Estadual de Goiás (UEG) pela infra-estrutura fornecida para realização do estudo, a meu orientador, a meus pais e ao Grupo de Química Teórica
Referências
1) L. Cullen, L. Kelly, S.O. Konnor, D.J. Fitzgerald, J. Pharmacol. Exp. Ther. 287(1998) 578. 2) Dubois R, Abramson S, Crofford L et al - Cyclooxigenase in biology and disease. Faseb J. 1998;12:1063-1088,. 3) Bhattacharya, K. Kankanala, S. Pal, A. K. Mukherjee a, Journal of Molecular Structure. 975 (2010) 40–46. 4) C. Michaux, C. Charlier, F. Julémont, X. de. Leval, J. –M. Dogné, B. Pirotte, F.Durant, Eur. J. Med. Chem. 40 (2005) 1316. 5) T. Inaba, K. Tanaka, R. Takeno, H. Nagaki, C. Yoshida, S. Takano, Chem. Pharm. Bull. 48 (2000) 131. 6) CAR & PARRINELLO. Phys. Rev. Lett., v. 55, n. 22, p. 2471-2474, 1985. 7) GAUSSIAN 09, Revision D.01, Frisch M. J. et al. Gaussian, Inc., Wallingford CT, 2009.
Patrocinadores







Apoio









Realização

