ISBN 978-85-85905-19-4
Área
Iniciação Científica
Autores
Cardoso Gomes, G. (UFPA) ; Leal Moreira, G. (UFPA) ; Nahum Alver, C. (UFPA) ; Lameira Silva, J. (UFPA)
Resumo
Determinação estrutural da proteína PhoR de C. pseudotuberculosis. Foi utilizado técnicas de modelagem molecular, dinâmica molecular e cálculo de energia livre, estes foram realizados em triplicata. Na modelagem molecular, foi obtido estruturas em cis e em trans, que apresentaram Ramachandran e LGscore de 91,9%, 5,021 e 94,4%, 4,068 respectivamente. As estruturas foram utilizadas na dinâmica molecular, com a conformação cis apresentando uma maior estabilidade e apresenta a coordenação com o íon de magnésio que está presente no complexo e é importante para a reação. Os resultados de energia para os sistemas foram -27,67 Kcal/mol em cis e 26,66 Kcal/mol em trans. Concluímos que a estrutura cis apresenta uma maior estabilidade e assim é a provável estrutura tridimensional para a proteína PhoR
Palavras chaves
Modelagem Molecular; Dinâmica Molecular; PhoR
Introdução
O conhecimento dos mecanismos moleculares envolvidos na resistência de bactérias patogênicas é importante para tratamentos mais direcionados das enfermidades causadas por estes microrganismos. A identificação de proteínas relacionadas à resistência bacteriana e o conhecimento das estruturas 3D apresentam grande importância para a compreensão das reações envolvidas nos processos bioquímico responsáveis pela resistência bacteriana. Sistemas Regulatório de dois componentes (TCS- do inglês Two Component System) são um dos principais mecanismos de detecção e resposta à mudanças no meio ambiente utilizado pelas bactérias. A bactéria C. pseudotuberculosis é causadora da linfadenite caseosa, uma enfermidade detectada mundialmente, mas com maior incidência na Austrália, Nova Zelândia, África do Sul, Estados Unidos e Brasil. O patógeno C. pseudotuberculosis possui 10 TCS, a proteína sensora histidina quinase PhoR, pertencente ao sistema regulatório PhoPR relacionado com a regulação de genes de virulência. PhoR é uma proteína trans-membranar ativada por uma reação de autofosforilação em um resíduo de histidina catalítica, está é a primeira reação no processo de ativação do TCS PhoPR. Entretanto, a porção citoplasmática dimérica desta proteína pode apresentar dobramento favorável para reação de cis ou trans-autofosforilação. Neste trabalho, a estrutura da proteína nativa PhoR de C. pseudotuberculosis foi determinada a partir de técnicas de modelagem molecular. Este modelo estrutural poderá ser usado para o desenho de novos inibidores deste alvo.
Material e métodos
Modelagem Comparativa: Primeiramente, foi identificado estruturas de proteínas conhecidas, que servirão de molde (template) para a determinação das estruturas das proteínas-alvo (target). As buscas por moldes foram feitas no PDB observando o alto score, resolução e identidade. Neste trabalho foram usados o programa Modeller versão 9v14, um dos programas mais empregados em modelagem molecular, e o servidor automatizado SWISS-MODEL. Dinâmica Molecular: Primeiramente, foi feito uma preparação no ATP o qual teve a sua carga e posição otimizadas através do programa gaussian09. Após, foi feito uma avaliação de protonação dos resíduos ionizáveis da proteína, sendo que para os resíduos de Glutamato e Aspartato foi feito também uma análise de posicionamento. Todos os aminoácidos foram avaliados levando em consideração o pKa utilizando o servidor PropKa. A histidina catalítica foi prontamente protonada baseando-se na literatura. O sistema foi preparado utilizado o modulo tleap, onde foram adicionados os hidrogênios ao sistema e posteriormente imerso em uma caixa octaédrica contendo moléculas de agua do tipo TIP3P em uma distância de 10 Å. Para as simulações foram usados os campos de forca AMBER ff99SB-ILDN na proteína e AMBER geral. Após, foi realizado a minimização do sistema, sendo minimizados primeiramente os hidrogênios, posteriormente as moléculas de agua e os contra íons e por último todos os átomos. Posteriormente, o sistema foi aquecido lentamente até a temperatura de 300 K e equilibrado. O último passo foi a produção para simular as interações da enzima durante um período de tempo. Os resultados foram avaliados usando o programa AMBER12 e o pacote AMBERTOOLS 13. Os cálculos de energia livre foram realizados no programa sietraj.
Resultado e discussão
O modelo para a conformação cis foi obtido utilizando a proteína sensor de osmolaridade EnvZ quimera de E. Coli (EnvZchim)(PDB:4KP4); e para a conformação trans foi obtido utilizando proteína sensor de osmolaridade EnvZ nativa de E. coli (PDB:4CTI). Ambos modelos foram avaliados utilizando os paramentos Ramachandran e LGscore e apresentaram valores de 91,9%, 5,021 e 94,4%, 4,068 respectivamente. O que mostra que ambos os modelos foram bem construidos e apresentam boa estrutura tridimensional. A figura 1 mostra a estrutura em cis(a) trans(b) e os alinhamentos das estruturas com os modelos, cis e 4KP4(c) e trans e 4CTI(d).
A dinâmica foi realizada tanto nos modelos gerados quanto nos moldes para que assim pudesse se ter uma analise de movimentação. As analises foram realizadas através do gráfico de RMSD que demonstra a movimentação da proteína pelo tempo. Na figura 2 podemos observar os resultados de RMSD pra cada sistema e replica, e possível observar que tanto as estruturas EnvZ(a) e EnvZchim(b) apresentam uma maior estabilidade durante a dinâmica, porem somente a conformação cis(c) apresenta estabilidade para a proteína PhoR, sendo que a conformação trans(d) se movimenta muito mais. E possível observar também que apenas em cis ocorre a correta coordenação com o íon magnésio(e) enquanto em trans isto não ocorre(f).
Os resultados de energia livre mostraram uma media de -27,67 Kcal/mol em cis e 26,66 Kcal/mol em trans, isto provavelmente e devido ao fato do método não levar em conta as aguas no sistema, as quais são importantes para a reação, porem mesmo assim podemos perceber que a reação em cis e mais espontânea que em trans.
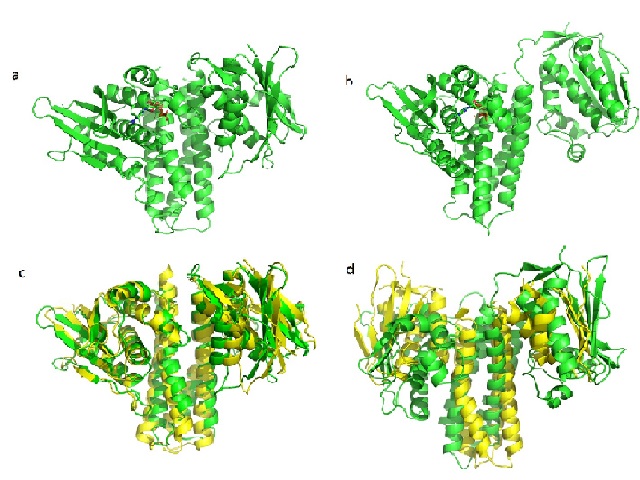
Estrutura em a)conformcacao em cis b) conformacao em trans c)alinhamento de cis com a EnvZchim e d) alinhamento de trans com a EnvZ
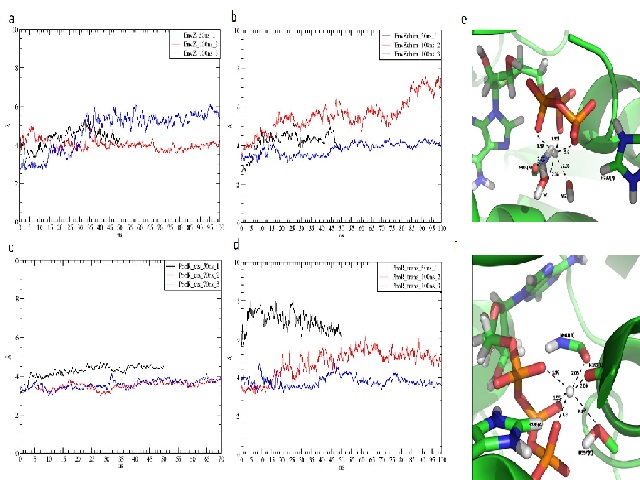
RMSD da a)EnvZ b)EnvZchim c)conformacao cis d)conformacao trans; coordenacao em e)cis e f) trans
Conclusões
Os resultados apresentados neste trabalho demonstraram que a conformação cis para o complexo PhoR é a conformação mais provável para a proteína, pois esta conformação apresenta uma menor movimentação, maior estabilidade do sitio catalítico e além disso apresenta um valor de energia favorável. Esta conformação também é a única a apresentar a coordenação com o íon magnésio o que está de acordo com a literatura para proteínas análogas a PhoR.
Agradecimentos
Ao LPDF ao qual eu participo. A UFPA onde sou aluno. Ao cnpq pelo fomento.
Referências
ALBERTS, B.; JOHNSON, A.; LEWIS, J. Molecular Biology of the Cell. 4th edition. New York: Garland Science; 2002.
ARNOLD, K.; BORDOLI, L.; KOPP, J.; SCHWEDE, T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modeling. Bioinformatics, v. 22, n.2 , p. 195-201, 2006
ARSENAULT, J.; GIRARD, C.; DUBREUIL, P.; DAIGNAULT, D.; GALARNEAU, J. R.; BOISCLAIR, J.; SIMARD, C.; BÉLANGER, D. Prevalence of and carcass condemnation from maedi–visna, paratuberculosis and caseous lymphadenitis in culled sheep from Quebec, Canada. Preventive Veterinary Medicine, v. 59, n.2, p. 67-81, 2003.
BERMAN, H. M.; WESTBROOK, J.; FENG, Z.; GILLILAND, G.; BHAT, T. N.; WEISSIG, H.; SHINDYALOV, I. N.; BOURNE, P. E. The Protein Data Bank. Nucleic Acids Research, v. 28, n. 1, p. 235-242, 2000
CASINO P.; MIGUEL-ROMERO L.; MARINA A. Visualizing autophosphorylation in histidine kinases. Nature Communications, v. 5, n. 3258-3258, 2014.
DORELLA, F. A.; PACHECO, L. G.; OLIVEIRA, S. C.; MIYOSHI, A.; AZEVEDO, V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Journal Veterinary Research, v. 37, n.2, p. 201-218, 2006.
ESWAR, N.; MARTI-RENOM, M. A.; WEBB, B.; MADHUSUDHAN, M. S.; ERAMIAN, D.; SHEN, M.; PIEPER, U.; SALI, A. Comparative Protein Structure Modeling Using MODELLER. Current Protocols in Bioinformatics, John Wiley & Sons, Inc., Supplement 15, 5.6.1-5.6.30, 2006
FONTAN, P.A.; WALTERS, S.; SMITH, I. Cellular Signaling Pathways and Transcriptional Regulation in Mycobacterium tuberculosis: Stress control and virulence. Current Science, v. 86, n.1, p. 122-134, 2004
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D.01: secondary title. Wallingford CT, 2009.
GUEX, N.; PEITSCH, M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, v. 18, n.15 , p. 2714–2723, 1997
HOCH, J.A. Two-component and phosphorelay signal transduction. Current Opinion in Microbiology, v. 3, n. 2, p. 165-170, 2000.
JORGENSEN, W. L.; CHANDRASEKHAR, J.; MADURA, J. D.; IMPEY, R. W.; KLEIN, M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemistry Physics, v. 79, n. 2, p. 926-935, 1983.
KURIA, J. K. N.; MBUTHIA, P. G.; KANG ETHE, E. K.; WAHOME, R. G. Caseous lymphadenitis in goats: the pathogenesis, incubation period and serological response after experimental infection. Veterinary Research Communications, v. 25, n. 2, p. 89-97, 2001
LI, H.; ROBERTSON, A. D,; JENSEN, J. H. . Very fast empirical prediction and rationalization of protein pK(a) values. Proteins: Structure, Function, and Bioinformatics, v. 61, n. 4, p. 704-721, 2005.
LINDORFF-LARSEN, K.; PIANA, S.; PALMO, K.; MARAGAKIS, P.; KLEPEIS, J. L.; DROR, R. O.; SHAW, D. E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins, v. 78, n. 8, p. 1950–1958, 2010.
MUNIZ, J. R. C. Aplicação da bioinformática nos estudos dos genes e enzimas envolvidos na síntese da goma fastidiana produzida pela Xylela fastidiosa. 2003. 124p. Dissertação (Mestrado) - Instituto de Física de São Carlos, Universidade de São Paulo, São Carlos. 2003.
PEEL, M. M.; PALMER, G. G.; STACPOOLE, A. M.; KERR, T. G. Human lymphadenitis due to Corynebacterium pseudotuberculosis: report of ten cases from Australia and review. Clinical Infectious Diseases, v. 24, n. 2, p.185-191, 1997
RASKO, D. A.; MOREIRA, C. G.; LI, R.; READING, N. C.; RITCHIE, J. M.; WALDOR, M. K.; WILLIAMS, N.; TAUSSIG, R.; SHUGUANG, W.; ROTH, M.; HUGHES, D.; HUNTLEY, J. F.; FINA, M. W.; FALCK, J. R.; SPERANDIO, V. Targeting QseC signaling and virulence for antibiotic development. Science, v. 321, n. 5892, p. 1078-1080, 2008.
ROE, D. R.; CHEATHAM, T. E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. Journal of Chemical Theory and Computation, v. 9, n. 7, p. 3084-3095, 2013.
STOCK, A. M.; ROBINSON, V. L.; GOUDREAU, P. N. Two-component signal transduction. Annual Review Biochemistry, v. 69, n. 1,p. 183–215, 2000
WANG, J. M.; WOLF, R. M.; CALDWELL, J. W.; KOLLMAN, P. A.; CASE, D.A. Development and testing of a general amber force field. Journal of Computer Chemistry, v. 25, n. 9, p. 1157-1174, 2011
CUI, Q.; SULEA, T.; SCHRAG, J. D.; MUNGER, C.; HUNG,M.-N.; NAÏM, M., CYGLER, M.; PURISIMA, E. O. Molecular Dynamics and Solvated Interaction Energy Studies of Protein-Protein Interactions: the MP1-p14 Scaffolding Complex. J. Mol. Biol, v. 379, n. 4, p. 787-802, 2008.
NAÏM, M.; BHAT, S.; RANKIN, K. N.; DENNIS, S.; CHOWDHURY, S. F.; SIDDIQI, I.; DRABIK, P.; SULEA, T.; BAYLY, C.; JAKALIAN, A.; PURISIMA, E. O. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J. Chem. Inf. Model, v. 47, n. 1, p. 122-133, 2007.
Patrocinadores



Apoio







Realização

