ISBN 978-85-85905-19-4
Área
Ambiental
Autores
Costa, W.A. (UFPA) ; Silva Filho, F.G.S. (IESAM) ; Oliveira, M.S. (UFPA) ; Carvalho Jr, R.N. (UFPA) ; Martelli, M.C. (UFPA) ; Brasil, D.S.B. (UFPA)
Resumo
Os hidrocarbonetos monoaromáticos são responsáveis pela contaminação de solos e rios. O método mais utilizado para sua remoção é a adsorção por carvão ativado. O objetivo deste trabalho é elucidar através de simulações de dinâmica molecular as interações entre as estruturas moleculares do carvão ativado e as do p-xileno. O modelo de simulação foi representado por duas moléculas de carvão ativado distanciadas de 20 Å e solução aquosa de p-xileno, o qual foi submetido a aquecimento até 298 K e dinâmica molecular durante 30 ns, com método semi-empírico. Constatou-se que os grupos oxigenados presentes na estrutura do carvão ativado conferem caráter ácido e carga superficial negativa à mesma e devido a isto a adsorção tornou-se viável uma vez que o poluente assume carga positiva.
Palavras chaves
Carvão ativado; Xileno; Dinâmica molecular
Introdução
Os hidrocarbonetos monoaromáticos encontram-se entre os principais contaminantes de águas subterrâneas. Estes contaminantes tais como benzeno, tolueno e xilenos (orto-, meta- epara-) são poderosos depressores do sistema nervoso central, apresentando toxicidade crônica, mesmo em pequenas concentrações (da ordem de μg.L-1) (SILVA et al, p. 2, 2002). Dentre os vários métodos de remoção destes poluentes, a adsorção por carvão ativado é o método mais utilizado, pois apresenta uma habilidade perfeita para adsorver componentes orgânicos de baixo peso molecular, como é o caso do p-xileno (SCHNEIDER, p. 15, 2008). As simulações computacional e dinâmica de sistemas moleculares estão indubitavelmente destinadas a tornar-se um meio cada vez mais importante para a investigação das estruturas (JORGENSEN et al, p. 1, 1998). Deste modo, neste trabalho, descreve-seuma abordagem prática da modelagem molecular como umaferramenta interessante para estudar as relações estrutura-atividade do carvão ativado sob análise, principalmente no que se refere à análise do processo de adsorção de p-xileno sobre a superfície do carvão.
Material e métodos
2.1. Obtenção das Estruturas e Construção dos Modelos Moleculares O modelo assumido para a representação molecular do carvão ativado foi proposto por COSTA (p. 58, 2014) (Figura 1a). O poluente usado na simulação, por sua vez, foi o p-xileno (Figura 1b). Esse modelo foi desenhado utilizando o software Marvin Scketch e otimizado por calculo de DFT, a nível de teoria BELYP e base 6-31G pelo programa Gaussian versão 9. Os arquivos obtidos da otimização foram usados para gerar os dados de coordenadas tridimensionais, cargas, ângulos e diedros de ligação e topologia, utilizando o código Antechamber, do pacote AMBER e campo de força FF99SB. 2.2. Dinâmica Molecular Dados de diâmetro de poro, diâmetro da molécula de cada um dos poluentes e da distância entre as placas foram obtidos a partir de dados da literatura e ferramentas de modelagem molecular (LIMA et al, p. 3, 2012; RAMOS, p. 55, 2012). O pacote AMBERTOOLS versão 13 foi utilizado para construção do modelo de mesoporo e do sistema de adsorção contendo carvão e poluentes (AMBER, 2013). Em seguida, o modelo foi parametrizado e solvatado com a ferramenta tleap e submetido a cálculos de minimização, aquecimento e dinâmica molecular. Foi usado o campo de força FF99SB. A etapa de aquecimento simulou o aumento de temperatura de 0 a 25ºC e foi dividido em cinco etapas com incremento de 5ºC e 500 ps de simulação em cada etapa. A etapa de dinâmica molecular simulou o estado do sistema em instantes de 0 a 30 ns dividido em cinco etapas com incremento de 2 ns em cada. Os resultados, uma representação tridimensional das condições impostas, são imagens obtidas e renderizadas pelo programa VMD (Figura 2a).
Resultado e discussão
A Figura 2b representa o estado final da dinâmica molecular omitindo as
moléculas de água, após 30 ns de cálculos.
A otimização geométrica altera a geometria molecular para diminuir a energia
do sistema e produz uma conformação mais estável (HYPERCHEM, p. 129, 2002;
LEACH, p. 273, 2001).
O modelo foi solvatado para que se tivesse a simulação de um ambiente aquoso
como ocorre em sistemas de adsorção reais.
A minimização de energia antes da realização da dinâmica molecular, além de
levar a estrutura para um mínimo de energia, também foi feita com o intuito
de remover qualquer “mau contato” criado pela solvatação (devido à
dissociação iônica das moléculas de água) (RAMOS, p. 66, 2012).
Grupos funcionais e elétrons deslocalizados são fatores que determinam o
caráter químico (ácido ou básico) da superfície do carvão ativado, uma vez
que o oxigênio pode estar presente sob várias formas. No caso da estrutura
do carvão sob análise, encontram-se os grupos éter, carbonila e lactona que
tornam a estrutura mais ácida, apolar e consequentemente, a mesma acaba
apresentando carga superficial negativa (FERNANDES, p. 30, 2005; LOPEZ-
RAMON et al, p. 1, 1999). Segundo WIBOWO et al (p. 4, 2006), carvões com
poucos
grupos ácidos oxigenados superficiais apresentam maior capacidade de
adsorção.
O caráter não polar da superfície no carvão ativado é fator preponderante na
adsorção de moléculas não polares, podendo ser incrementada pela adequada
modificação da natureza química da superfície do carvão (por exemplo:
tratamento com ácidos) (YANG, p. 96, 2003).
Devido à sua não-polaridade e por possuir um volume maior de microporos, o
carvão adsorve mais fortemente as moléculas orgânicas apolares ou fracamente
polares, como é o caso do p-xileno (YANG, p. 79, 2003).
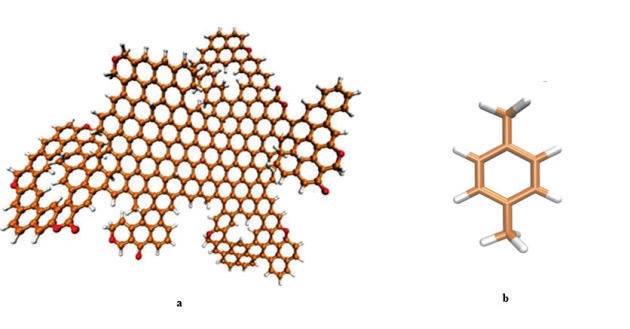
Modelo tridimensional do carvão ativado proposto (a); Modelo estrutural do p-xileno (b)
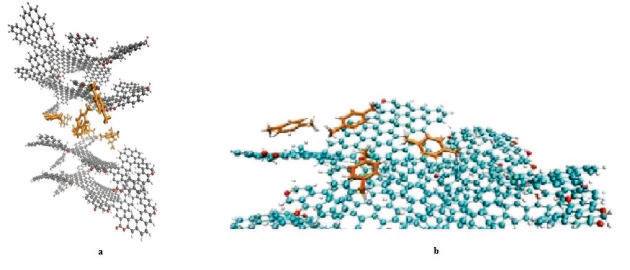
Modelo com as moléculas de p-xileno inseridas (a); Modelo após 30 ns de dinâmica molecular (b)
Conclusões
Verificou-se que a estrutura de carvão utilizada apresentou boa capacidade de adsorção de p-xileno em soluções aquosas, o que se dá principalmente devido ao caráter apolar da estrutura, caracterizado pelos grupos funcionais identificados na mesma.
Agradecimentos
À UFPA, CAPES e CNPQ.
Referências
AMBER MD. Introductory Tutorials, disponível em <http://ambermd.org/tutorials/>. Acesso em: 20/02/2013
COSTA, W. A. Simulação Computacional da Adsorção dos Poluentes Benzeno, Tolueno e p-Xileno Sobre Carvão Ativado. 2014. 89 f. Dissertação (Mestrado em Engenharia Química) – Instituto de Tecnologia, Universidade Federal do Pará, Belém, 2014.
FERNANDES, R. Adsorventes Alternativos para Remoção de Fenol em Solução Aquosa. 2005. 91 f. Dissertação (Mestrado em Engenharia Química) – Centro Tecnológico, Universidade Federal de Santa Catarina, Florianópolis, 2005.
HYPERCHEM. HyperChem Release 7 for Windows – Getting Started. Tools for Molecular Modeling. USA: HyperCubeInc, 2002.
JORGENSEN, W. L.; TIRADO-RIVES, J. The OPLS Potential Functions for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. Journal of the American Chemical Society. Volume 10, number 6, march 16, 1998.
LEACH, A. R. Molecular Modelling, Principles and Applications. 2nd edition. Dorchester (UK): Pearson Education Limited, 2001. 773 p.
LIMA, A. E. O.; MARIA, F. C. de; GOMES, V. A. M.; OLIVEIRA, J.C.A; LUCENA, S. M. P. de. Estudo da Adsorção de CO2 em Materiais Carbonosos Impregnados Via Simulação Molecular. In: XIX CONGRESSO BRASILEIRO DE ENGENHARIA QUÍMICA, 2012, Buzios, 2012.
LOPEZ-RAMON, M.V.; STOECKLI, F.; MORENO-CASTILLA, C.; CARRASCO-MARIN,F. On the Characterization of Acidic and Basic Surface Sites on Carbons by Various Techniques. Carbon 37 (1999) 1215–1221.
RAMOS, R. M. C. Determinação Computacional dos Efeitos da Mutagénese em Interfaces Proteína-DNA. 2012. 155 f. Dissertação (Mestrado em Química) - Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade do Porto, Porto, 2012.
SCHNEIDER, E. L. Adsorção de Compostos Fenólicos Sobre Carvão Ativado. 2008. 96 f. Dissertação (Mestrado em Engenharia Química) – Centro de Engenharias e Ciências Exatas, Universidade Estadual do Oeste do Paraná, Toledo, 2008.
SILVA, R. L. B.; BARRA, C. M.; MONTEIRO, T. C. N.; BRILHANTE, O. M. Estudo da Contaminação de Poços Rasos por Combustíveis Orgânicos e Possíveis Consequências para a Saúde Pública no Município de Itaguaí, Rio de Janeiro, Brasil. Cad. Saúde Pública, Rio de Janeiro, 18(6):1599-1607, nov-dez, 2002.
WIBOWO, N.; SETYADHI, L.; WIBOWO, D.; SETIAWAN, J.; ISMADJI, S. Adsorption of Benzene and Toluene from Aqueous Solutions onto Activated Carbon and its Acid and Heat Treated Forms: Influence of Surface Chemistry on Adsorption. Journal of Hazardous Materials. 146 (2007) 237–242.
YANG, R. T. Adsorbents: Fundamentals and Applications. U.S.A.: John Wiley & Sons, 2003. 425 p.
Patrocinadores



Apoio







Realização

