Avaliação de Derivados N,N-Dibenzilados de α-Aminoácidos Naturais como Novos Organocatalisadores Quirais.
ISBN 978-85-85905-21-7
Área
Química Orgânica
Autores
Meirelis, F.P. (UFRJ - IPPN) ; Pereira, V.L.P. (UFRJ - IPPN) ; Torres, E.R.B. (IFRJ - MARACANA)
Resumo
Neste trabalho, visamos sintetizar novas moléculas quirais, baseadas em 1,3- dióis, a partir de (L)-aminoácidos naturais e avaliar seu uso como organocatalisadores quirais. A estrutura deverá conter sítios básicos com força suficiente para promover desprotonações em substratos adequados (por exemplo nitroalcanos) e ligar-se aos reagentes pró-quirais através de ligações de hidrogênio e interações pi-pi (“π stacking force”). Os resultados preliminares mostraram que o sistema básico KF 1 equiv/iPrOH (entrada 3, esquema 4), foi mais adequado (53% de rendimento) para produzir o intermediário-chave 2 via a reação nitroaldólica entre o aldeído 4a e o nitroálcool 3a.
Palavras chaves
Reação Nitroaldólica; Reação de Michael; Simetria C2
Introdução
A catálise orgânica enantiosseletiva apesar de ser conhecida desde o início da década de 70 (HAJOS et al., 1974), somente emergiu como uma das mais importantes estratégias sintética para a obtenção de substâncias enantiomericamente enriquecidas em elevados excessos enantioméricos em em 2000, com o trabalho de LIST et al., 2000. Esta estratégia apresenta inúmeras vantagens sobre as que utilizam indutores de quiralidade ligado covalentemente ao substrato pró- quiral, como a "chiron aproach", a catálise enantiosseletiva via metais de transição mais ligante quiral e a transformação enzimática e microbiológica. Dentre as vantagens destacam-se o uso de quantidades sub- estequiométricas do organocatalisador quiral e a não utilização de metais de transição que além de tóxicos, atuam geralmente promovendo vários equilíbrios químicos difíceis de serem controlados, o que ocasiona em muito casos, uma difícil reprodutibilidade nos excessos enatioméricos já estabelicidos. No caso dos organocatalisadores quirais o processo de ligação da molécula quiral ao substrato pró-quiral dá-se através de ligações secundárias tais como ligação de hidrogênio, interações pi-pi ou "π- stacking force” (IVERSON et al.,2012), interações eletrostáticas cátion- ânion e também ligações covalentes temporárias. Na maioria das vezes as reações organocatalisadas não necessitam de atmosfera inerte, condições anidras e temperaturas elevadas e têm sido utilizadas na preparação de intermediários sintéticos avançados, biomoléculas, fármacos e produtos naturais complexos em elevados excessos enantioméricos (LIST et al., 2000). Propomos sintetizar novas moléculas quirais, baseadas no cerne 1,3-dióis, a partir de (L)-aminoácidos naturais e avaliar seu uso como organocatalisadores quirais. A estrutura deverá conter sítios básicos com força suficiente para promover desprotonações em adequados substratos (nitroalcanos) e ligar-se aos reagentes pró-quirais através de ligações de hidrogênio e interações pi-pi (“ ¶-stacking force”).
Material e métodos
Através da retroanálise proposta (esquema 1), o potencial catalisador orgânico quiral 1, possuidor de um eixo de simetria C2 e de uma porção 1,3-diol, poderia ser sintetizado via desnitração clássica de 2 promovida por Bu3SnH catalisada por AIBN, em tolueno. O nitroálcool 2 viria da reação nitroaldólica (reação de Henry) entre o nitroálcool anti-3a e o aldeído 4a. Já anti- 3a será sintetizado por reação anti-diastereosseletiva por tratamento de 4a com nitrometano, sob catálise básica promovida por TBAF.3H2O (PEREIRA). Por sua vez o alfa-alaninaldeído 4a e seu análogo 4b) serão produzidos a partir do correspondente álcool via oxidação promovida pelo reagente de Swern. Já os álcoois 7a e 7b serão produzidos via redução por LiAlH4 dos ésteres tribenzilados 5a, b. Estes últimos são produzidos via perbenzilação do aminoácidos naturais alfa-(L)-fenilalanina (6a) e em alfa-(L)-Leucina (6b) em BnBr/K2CO3-EtOH. A metodologia descrita a seguir foi obtida a partir das melhores condições reacionais testadas e descritas nos resultados e discussões.
Resultado e discussão
Iniciamos o trabalho sintetizando o nitroálcool 3a, precursor do alvo
1, e os N,N-tribenzilados éster 5b e do álcool
7b, em rendimentos superiores a 90,0%, utilizando rota otimizada em
nosso grupo de pesquisas, esquema 2. Os intermediários 5b e
7b, foram avaliados quanto a capacidade de seus grupos N,N-
dibenzilas agirem como sítios catalíticos básicos promovendo a desprotonação
do 2-nitropropano e concomitantemente induzirem a enantiosseletividade no
produto 1,3-dinitrado 9, proveniente da reação de Michael com o
nitroestireno 8,esquema 3.
Verificamos que 5b e 7b promoveram a reação de Michael via
formação do ânion nitronato e adição ao nitroestireno 8 em
rendimentos de 68,0 e 22,0 %, respectivamente. As substâncias 5b e
7b foram capazes de gerar o íon nitronato do 2-nitropropano, porém
não houve qualquer indução de quiralidade no aduto de Michael 9, como
evidenciado pelo uso de CLAE, utilizando coluna cromatográfica quiral.
Constatado a basicidade do grupo N,N-dibenzila, partimos para a
síntese do potencial organocatalisador com eixo C2. Assim, fizemos reagir
4a com 3a sob a ação de diversos sistemas catalíticos básicos,
esquema 4.
Dentre os sistemas básicos estudados, KF/iPrOH mostrou ser mais eficiente
para a promoção da reação nitroaldólica (entrada 3, esquema 4). O
nitro-1,3-diol derivado 2 foi obtido na faixa de 45,0 – 53,0% de
rendimento como uma mistura de quatro diastereoisômeros (2,2:1,7:1,5:1,0,
entrada 3, esquema 4) como evidenciado pela análise de RMN-13C e RMN-
1H. O sinal que absorve em entre 92,0 – 98,0 ppm no espectro de RMN-13C
(CH-NO2) inequivocamente mostra a formação de 2.
A desnitração de 2 promovida por Bu3SnH é catalisada pelo
iniciador de radicais livres AIBN (bis-AzoIsoButiroNitrila) e deverá
produzir 2, como uma mistura de dois diastereoisomeros em C3,
passíveis de separação via cromatografia em coluna de gel de sílica. Estes
terão a sua ação como organocatalisador enantiosseletivo investigada em
reações de Michael de 8 com 2-nitropropano.
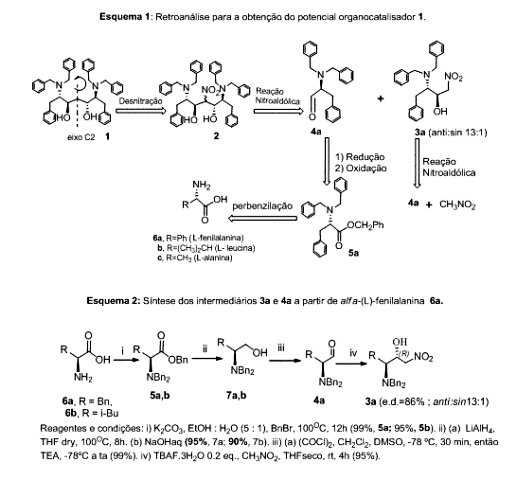
Esquema 1: Retroanálise para obtenção do potencial organocatalisador 1. Esquema 2: Síntese dos intermediários 3a e 4a a partir de 6a.
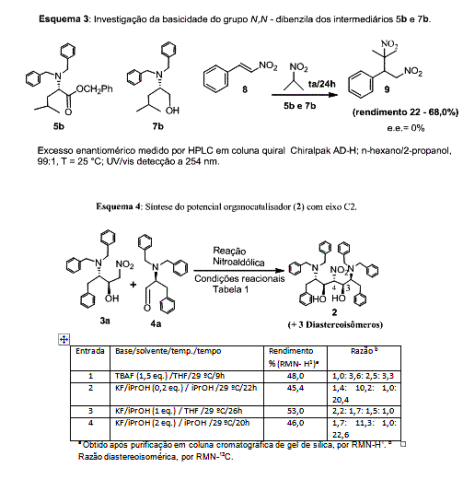
Esquema 3:Investigação da basicidade do grupo N,N-dibenzila dos intermediários 5b e 7b. Esquema 4: Síntese do potencial organocatalisador 2 (eixo C2).
Conclusões
Os pares de elétrons do grupo N,N-dibenzila de 5b são suficientemente básicos para desprotonar o nitropropano e catalisar uma reação de Michael. A reação nitroaldólica entre 3a e 4a, catalisada pelo ânion fluoreto é possível e produz 2 diastereosseletivamente. Novas condições reacionais estão sendo testadas a fim de aumentar o rendimento e distereosseletividade. A desnitração de 2 deverá produzir 1 como uma mistura diastereoisomérica de 2,0:1,0. Após separação dessa mistura em coluna cromatográfica, os testes da ação catalítica e da indução de quiralidade na reação de 8 com nitropropano poderão ser iniciados.
Agradecimentos
Agradeço a CAPES pela bolsa de Doutorado.
Referências
HAJOS, Z. G. et al;. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615
IVERSON, B. L. et al Rethinking the term ¶-stacking Chem. Sci., 2012,3, 2191-2201.
LIST, B., BARBAS III et al. Proline-Catalyzed Direct Asymmetric Aldol Reactions J. Am. Chem. Soc. 2000, 122, 2395.
LIST, B.; Introduction: Organocatalysis Chem. Rev. 2007, 107, 5413.
PEREIRA, V. L. P. et al Beilstein. A versatile and efficient approach to the synthesis of chiral 1,3-nitroamines and 1,3-diamines via conjugate addition to new (S,E)-γ-aminated nitroalkenes derived from L-α-aminoacids. J. Org. Chem. 2013, 9, 832–837.































