Síntese e avaliação citotóxica in vitro de novas 2-amino-4-fenil-pirimidinas substituídas por carbo/heterociclos aromáticos com atividade antineoplásica
ISBN 978-85-85905-21-7
Área
Química Orgânica
Autores
Pina, J.W.S. (UFES) ; Loureiro, L.B. (UNICAMP) ; Fernandes, M.R.N. (UNICAMP) ; Ferreira, C.V. (UNICAMP) ; Greco, S.J. (UFES)
Resumo
Neste trabalho foi sintetizado o 2-fenil-2H-1,2,3-triazol-4-carboxaldeído em rendimentos satisfatórios. Em seguida utilizou-se este aldeído e o p-anisaldeído para sintetizar chalconas substituídas através da metodologia clássica de condensação de Claisen-Schimidt que serviram de material de partida na síntese das 2-amino-4-fenil-pirimidinas substituídas no carbono C6 através da reação de Biginelli modificada por Atwal. As pirimidinas sintetizadas foram testadas quanto a sua atividade antitumoral através de estudos biológicos in vitro em linhagem de melanoma metastático humano (SKMEL-103). A atividade biológica mostrou claramente um sinergismo entre os núcleos pirimidínicos e triazol, pois, a moléculas contendo os dois núcleos foi aproximadamente três vezes mais potente.
Palavras chaves
Pirimidina; Reação de Biginelli; Atividade Anticâncer
Introdução
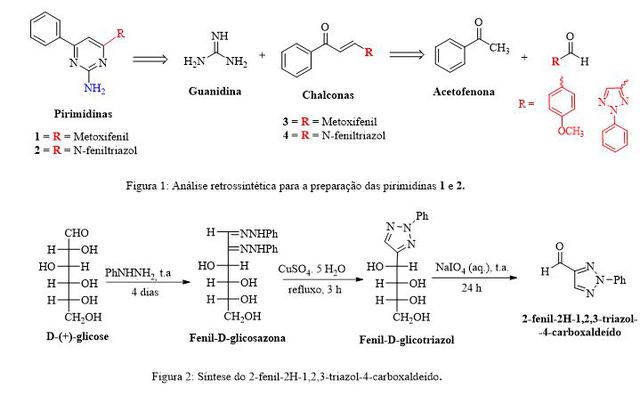
O panorama epidemiológico atual do câncer comprova a necessidade de estudos profundos e contínuos, uma vez que a doença contribui de maneira significativa para o aumento da morbimortalidade em todo o mundo. Segundo a Organização Mundial de Saúde (OMS) em um informativo publicado em fevereiro de 2015, cerca de 14 milhões de novos casos e 8,2 milhões de mortes estão relacionados ao câncer em 2012. A previsão é de que nas próximas duas décadas, este número aumente em 70%, ou seja, cerca de 24 milhões de novos casos. Dentre as principais causas mundiais de morte por câncer, um terço está relacionado a fatores comportamentais e de alimentação, como falta de atividade física, obesidade, quantidades ínfimas de vegetais ingeridos diariamente, utilização de álcool e tabaco. De todas essas, o hábito de fumar constitui o fator de risco que mais colabora para desenvolvimento do câncer, causando cerca de 20% de todos os cânceres e 70% do câncer de pulmão.(1) A terapia do câncer é baseada na cirurgia, radioterapia, que são, quando possível, bem-sucedidas intervenções regionais e na quimioterapia sistemática. Aproximadamente metade dos pacientes com neoplasia maligna não são curados por esses tratamentos e podem obter somente uma sobrevida prolongada ou até mesmo nenhum benefício considerável. O objetivo da maioria dos fármacos quimioterápicos atualmente em uso clínico é matar as células tumorais malígnas mediante inibição de algum mecanismo presente na divisão celular. Portanto, as drogas antitumorais desenvolvidas através dessa abordagem são citostática ou citotóxica. Entretanto, o avanço no conhecimento da biologia do tumor, nas décadas passadas, pode abrir caminho para o desenvolvimento de drogas mais potentes e mais específicas.(2) Fármacos específicos têm como objetivo o acúmulo preferencial princípio ativo no alvo celular independentemente do método e rota de administração.(3) Apesar do DNA continuar sendo um alvo essencial para a quimioterapia do câncer, esforços recentes tem sido direcionado para a descoberta de drogas antitumorais especificamente adequada para alvos moleculares aberrantes que são específico das células tumorais.(4) Essa nova geração de agentes antitumorais é baseado na pesquisa de processos de sinalização celular, angiogênese e metástase e na inibição de enzimas que, como a telomerase, são reativadas na maioria das células tumorais.(5) Para este fim, pode-se usar drogas que são pequenas moléculas ou outras estruturas macromoleculares, como anticorpos monoclonais que se ligam a antígenos presentes preferencialmente ou exclusivamente nas células do tumor, ligando-se diretamente a dupla hélice do DNA e consequentemente inibindo a transcrição.(6) Nesse sentido, a sinalização celular, que é o mecanismo pelo qual sinais que estão presentes no meio extracelular são transmitidos e interpretados pelas células, propicia alvos terapêuticos altamente seletivos. Nas células malignas, a ativação exacerbada dessas vias promove a proliferação e sobrevivência celular, sendo que as vias mais comumente ativadas são aquelas onde estão envolvidas proteínas quinases.(7) As pirimidinas, compostos importantes na inibição das quinases e por consequência em suas respectivas vias de sinalização, são heterociclos aromáticos de seis membros com dois nitrogênios no anel, separados entre si por um átomo de carbono.(8) Possui ocorrência natural, estando presente em nucleotídeos e na vitamina B1. O anel pirimidínico é uma unidade de construção do DNA e do RNA e por isso, arquiteturas químicas baseadas neste heterociclo exibem diversas atividades farmacológicas, tais como: atividade antifúngica, antibacteriana, antimicrobiana, anti-inflamatória, antiviral, anticâncer, antioxidante, antituberculose, antiparkinson, analgésica, antimalária, diurética, cardiovascular e anti-HIV.(9,10) Os compostos constituídos em seu arcabouço por pirimidinas podem atuar nas células de câncer por diferentes e variados mecanismos de ação, como por exemplo, inibição das enzimas fosforilase/nucleosidase (inibidores de PNP, MTAP e MTAN), inibição de tirosinas quinases (inibidores de HER2K, EGFRK, VEGFR2K, JAK), inibição de Fosfatidilinositol 3-quinases (inibidores de PI3K), inibição da histona deacetilase, inibição das proteínas serina/threonina quinases (inibidores de GCN2, mTOR e RAF), inibição da proteína Heat Shok 90 (Hsp90), inibição da autotaxina (Ats) e por fim, via inibição da proteína quinase de dupla-especificidade (inibidores de MPSI).(11) Diante do exposto nesse trabalho foram sintetizadas as novas 2-amino-4-fenil- pirimidinas substituídas por carbo/heterociclos aromáticos no carbono C6 1 e 2 com potencial atividade anticâncer através da reação de Biginelli modificada por Atwal entre chalconas e a guanidina.(12) As chalconas 3 e 4 foram, por sua vez, sintetizadas mediante a reação clássica de Claisen-Schimidt(13) usando para isso a acetofenona e os respectivos aldeídos não enolizáveis conforme mostrado na Figura 1.
Material e métodos
- Metodologia: As análises de cromatografia em camada fina foram realizadas utilizando cromatofolhas de alumínio recobertas com sílica gel UV254 (250 µm, 20x20 cm). As análises de espectroscopia de infravermelho foram realizadas utilizando o espectrômetro Perkin Elmer Spectrum 400. Todas as análises foram feitas utilizando o modo de Reflectância Total Atenuada (Attenuated total reflection - ATR) com cristal horizontal de seleneto de zinco (ZnSe). As análises de espectroscopia de RMN de ¹H e de ¹³C foram realizadas utilizando o espectrômetro Varian 400MHz e sonda de 5 mm Broadband 1H/X/D. Para a avaliação da citoxicidade foi realizada a análise de viabilidade celular por meio do ensaio do corante MTT (3-(4,5-dimetiltiazol-2-il)2,5-difenil brometo de tetrazólio - SIGMA) que é fundamentado na redução do MTT a uma formazana pelas mitocôndrias das células vivas. Assim, ao término das 24h de tratamento as células foram lavadas com 200 mL/poço de tampão fosfato (pH=7,0) e em seguida, foi adicionado 100 mL/poço de DMEM sem SFB, contendo 0,5 mg/mL de MTT. Posteriormente, as placas foram agitadas por 10 min em placa agitadora e as absorbâncias correspondente a cada poço foram obtidas em leitor de placas (ELx 800 BIO-TEK) em λ=570nm. Os valores das absorbâncias foram expressos como porcentagens de redução do MTT em relação ao controle negativo (%CTL).(14) A síntese do 2-fenil-2H-1,2,3-triazol-4-carboxaldeído foi realizada em três etapas a partir da D-(+)- glicose conforme descrito na literatura.(15) As chalconas 3 e 4 foram preparadas seguindo a metodologia geral descrita a seguir: a um balão de fundo redondo de 50 mL foram adicionados 3,1 mL de uma solução aquosa de NaOH 10% (m/m), 18 mL de metanol e 1 mmol do respectivo aldeído. Essa mistura foi mantida sob agitação e baixa temperatura (0-10°C) enquanto se adicionou, lentamente, 1 mmol da respectiva cetona. Ao término da adição, o banho de gelo foi retirado e a mistura reacional foi mantida a temperatura ambiente por tempos reacionais de 12h para 3 e 21h para 4. Ao término da reação, verificada por CCF, foi vertido de água destilada gelada e o sólido formado foi filtrado e lavado com água gelada. As pirimidinas 1e 2 foram preparadas conforme procedimento geral descrito a seguir: em um balão de fundo redondo de 10 mL adicionou-se 5 mL de etanol, 1,5 mmol de cloreto de guanidínio e 4 mmol de NaOH. Essa mistura foi mantida sob agitação em temperatura ambiente por cerca de 1h, adicionando-se, em seguida, 1 mmol da chalcona correspondente e elevando a temperatura até o refluxo do solvente (aproximadamente 80°C). O curso reacional foi monitorado por CCF e o término da reação foi verificado após um tempo de refluxo de 8h para 1 e 5 horas para 2. O sólido formado foi filtrado com funil analítico e lavando abundantemente com metanol e água, ambos gelados.
Resultado e discussão
Esse trabalho teve início através da síntese do 2-fenil-2H-1,2,3-triazol-4-
carboxaldeído, único aldeído utilizado nesse trabalho que não é comercial. A
preparação em três etapas foi realizada a partir da D-(+)- glicose, conforme
mostrado na Figura 2 e descrito na literatura.(15)
Uma vez obtidos todos os aldeídos realizou-se a síntese das chalconas 3 e 4
através da metodologia clássica de condensação de Claisen-Schimidt, na qual se
utilizou uma cetona enolizável, a acetofenona, na presença de hidróxido de sódio
aquoso e do respectivo aldeído não-enolizável (anisaldeído e o 2-fenil-2H-1,2,3-
triazol-4-carboxaldeído previamente sintetizado) em metanol como solvente.(13) A
chalcona 3 foi obtida como um sólido amarelo em 75% de rendimento e ponto de
fusão igual a 113-115 °C. Já o composto 4 foi obtido como um sólido bege em 93%
de rendimento e ponto de fusão igual a 131-133 °C.
As pirimidinas 1 e 2 foram sintetizadas através da reação de Biginelli
modificada por Atwal usando como material de partida as chalconas 3 e 4
previamente sintetizadas e o cloreto de guanidínio em meio básico de hidróxido
de sódio usando etanol como solvente, sob condições de refluxo.(14) A pirimidina
1 foi obtida como um sólido amarelo-claro em 91% de rendimento e ponto de fusão
igual a 163-164 °C. Já a pirimidina feniltriazólica 2 foi sintetizada em 70% de
rendimento como um sólido amarelo e ponto de fusão igual a 207-209 °C.
Os espectros de IV desses compostos mostraram as seguintes bandas
características: para a pirimidina 1 - 3359 e 3323 cm-1 referentes ao grupo NH2
ligado ao anel pirimidínico; banda em 1561 cm-1 relativa a estiramento da
ligação C=N e em 1237 cm-1 correspondente ao estiramento da ligação C-N; bandas
em 1258 e 1029 cm-1 relacionadas à ligação C-O; e ainda bandas em 1640 e 1359
cm-1 que são absorções características do anel pirimidínico. Já para 2 foram
descritas as bandas características do anel pirimidínico (1662 e 1346 cm-1),
bandas referentes a estiramentos C=N e C-N respectivamente em 1583 e 1227 cm-1,
além das bandas em 3475 e 3313 cm-1 referentes ao grupo NH2.
O espectro de RMN de ¹H da molécula-alvo 1 apresentou um único sinal na região
alifática como sendo um singleto, em 3,80 ppm, relativo aos hidrogênios da
metoxila ligada ao anel aromático. Na região mais desblindada do espectro
destacam-se ainda dois singletos: um em 6,61 ppm, alargado, que corresponde aos
hidrogênios da amina primária ligada à pirimidina; e outro em 7,61 ppm relativo
ao único hidrogênio presente no anel pirimidínico, sinal essencial à confirmação
da ocorrência da reação e formação do heterociclo pirimidínico. Os demais
hidrogênios aromáticos foram atribuídos com auxílio da espectroscopia de RMN de
correlação bidimensional 1H-1H (COSY). O espectro de RMN de 1H de 2 foi fácil
distinguir um singleto alargado em 6,92 ppm, integrando para dois hidrogênios,
que corresponde aos hidrogênios do grupamento amina. Há ainda neste espectro
dois singletos importantes: um em 7,75 ppm referente ao único hidrogênio do anel
pirimidínico e outro em 8,59 ppm referente ao hidrogênio triazólico, sendo este
o hidrogênio mais desblindado da molécula. A atribuição dos hidrogênios dos
grupos fenilas também foi feita após análise do 1H-1H (COSY).
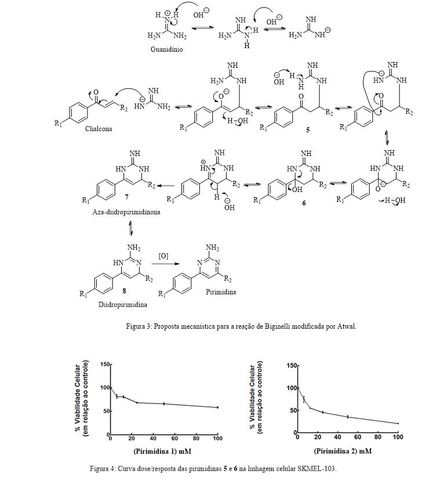
O mecanismo proposto para a reação de Atwal usando chalcona e guanidina é
descrito a seguir (Figura 3). A reação se inicia pela adição da guanidina na
chalcona, via adição conjugada como sugerido por Atwal(12) e discutido por El-
Rayyes(16) e por Svetlík.(17) A adição da guanidina desprotonada leva a formação
do intermediário 5 passando por uma etapa de equilíbrio ceto-enólico, na qual a
formação da forma ceto é termodinamicamente favorecida. Em seguida ocorre ataque
nucleofílico do outro nitrogênio da guanidina à carbonila da chalcona, formando
o intermediário hidroxilado 6. Essa hidroxila é eliminada via assistência
anquimérica do par de elétrons não ligante do nitrogênio adjacente, facilitando
a formação do intermediário 7 que é uma aza-diidropirimidinona. O composto 7
está em equilíbrio tautomérico com a diidropirimidina 8, sendo esta última a
forma favorecida como comprovado por Wendelin(18) ao isolá-lo. Este
intermediário, entretanto, é facilmente oxidado em solução, levando a formação
da pirimidina correspondente.
Estudos in vitro de MTT com as pirimidinas 1 e 2 foram realizados utilizando uma
linhagem celular tumoral humana: SKMEL-103 (melanoma metastático), que possui
alta agressividade e resistência a quimioterápicos convencionais. Ao se analisar
os resultados obtidos para cada uma das moléculas (Figura 4), foi possível
observar que a pirimidina 2 induziu uma diminuição da viabilidade celular de
SKMEL-103 com uma relação dose dependente, enquanto o aumento da dosagem da
pirimidina 1 não produziu efeitos significativos na viabilidade celular da
linhagem selecionada. Os valores de IC50 foram determinados e para a pirimidina
1 foi igual a 140,2 μM enquanto que para a pirimidina triazólica 2 foi de 34,33
μM. Além disso, foi evidenciado que apenas a última apresentou toxicidade
significativa na linhagem tumoral estudada e que nenhuma das duas apresentou
citotoxicidade frente a células saudáveis.
Estes resultados comprovam a ideia do sinergismo dos núcleos pirimidínico e
triazólico na atividade anticâncer, visto que a molécula que continha os dois
núcleos teve melhor desempenho, sendo quase três vezes mais potente que a
pirimidina 1.
Conclusões
As metodologias de síntese adotadas neste trabalho possibilitaram a preparação das chalconas 3 e 4 e das pirimidinas 1 e 2 propostas com rendimentos de bons a excelentes, todas caracterizadas por espectroscopia de infravermelho e de ressonância magnética nuclear de hidrogênio (RMN de 1H) e sua respectiva correlação bidimensional (COSY). O estudo biológico mostrou citotoxicidade da pirimidina 2 frente as células tumorais SKMEL-103 e ausência de toxicidade frente a células saudáveis, evidenciando sua ação seletiva. A pirimidina 1 não apresentou toxicidade relevante frente a linhagem de câncer humano estudada. O sinergismo entre os núcleos pirimidínico e triazólico foi comprovado através dos estudos biológicos que indicaram que a molécula contendo os dois núcleos 2 foi quase três vezes mais potente que a molécula que continha apenas o núcleo pirimidínico 1.
Agradecimentos
Os autores agradecem aos órgãos FAPES, CAPES e CNPq pelo apoio financeiro ao trabalho desenvolvido, e ao NCQP/DQUI/UFES pelo suporte técnico.
Referências
1. CANCER: Fact sheet n°297. Disponível http://www.who.int/mediacentre/factsheets/fs297/en/.
2. Bradburry, R. H.; Cancer in “Topics in Medicinal Chemistry”, Vol. 1; Springer-Verlag, Berlin Heidelberg.
3. Torchilin, V. P. Eur. J. Pharm. Sci. 2000, 11, 81.
4. Longley, D. B.; Harkin, D. P.; Johnston, P. G. Nat. Rev. Cancer 2003, 3, 330.
5. Nam, N-H; Parang, K. Curr. Drug Targets 2003, 4, 159.
6. Segota, E.; Bukowski, R. M. Cleve. Clin. J. Med. 2004, 71, 551.
7. Sos, M.L. et al. PNAS 2009, 106(43),18351.
8. Merugu, R.; Garimella, S.; Balla, D.; Sambaru, K. International Journal of PharmTech Research. 2015, 8(6), 88.
9. Yerragunta, V.; Patil, P.; Anusha, V.; KumaraSwamy, T.; Suman, D.; Samhitha, T. PharmaTutor. 2013, 1(2), 39.
10. Alodeani, E. A.; Izhari, M. A. e Srshad, M. Europ. J. Biom. Pharm. Sci. 2014, 1(3), 504.
11. a) Ramandeep, K.; Prabhkirat, K.; Sahil, S.; Gurpreet, S.; Samir M.; Preet, M.; Bedia, S.; Kunal, N. Recent Patents on Anti-Cancer Drug Discovery 2015, 10, 23; b) Koroleva, E. V.; Ignatovich, Z. I.; Sinyutich, Y. V.; Gusak, K. N. Russian J. Org. Chem. 2016, 52(2), 139.
12. Atwal, K. S.; Rovnyak, G. C.; O'Reilly, B. C.; Schwartz, J. J. Org. Chem. 1989, 54, 5898.
13. Suwito, H.; Kristanti, A.; Puspaningsih, N. J. Chem. Pharm. Res. 2014, 6, 1076.
14. Denizot, F.; LANG, R. J. Immun.l Meth. 1986, 89(2), 271.
15. Yejella, R. P.; Atla, S. R. Chem. Pharm. Bull. 2011, 59(9), 1079.
16. El-Rayyes, N. R. J.l Heteroc. Chem. 1982, 19, 415.
17. Svetlik, J.; Sallai, L. J. Heteroc. Chem. 2002, 39, 363.
18. Wendelin, W.; Schermanz, K. J. Heteroc. Chem. 1984, 21, 65.































