ESTUDO ELETRÔNICO/ESTRUTURAL DO FÁRMACO SINTÉTICO TAVABOROLE: UMA ABORDAGEM QUÂNTICA SEMI-EMPÍRICA
ISBN 978-85-85905-21-7
Área
Iniciação Científica
Autores
Bezerra, L.L. (UNIVERSIDADE ESTADUAL DO CEARA) ; Marinho, M.M. (UNIVERSIDADE FEDERAL DO CEARÁ) ; Marinho, E.S. (UNIVERSIDADE ESTADUAL DO CEARA)
Resumo
Tavaborole é um fármaco de origem sintética, que realiza o tratamento da onicomicose, apresentando vários efeitos colaterais. O objetivo desse trabalho foi utilizar rotinas de simulação computacional, configuradas para realizar cálculos quânticos semi-empíricos (PM7) para caracterizar o fármaco sintético Tavaborole, estruturalmente e eletronicamente, obtendo sua estrutura conformacional de menor energia, mais estável, identificar os possíveis sítios reacionais com maior densidade eletrônica (Flúor), gerando uma região com caraterísticas nucleofílicas. Foram plotados os orbitais moleculares de fronteira, que serão fundamentais para futuros cálculos de descritores moleculares, concluindo assim, que este trabalho constitui uma etapa inicial para o melhoramento do fármaco (Drug designer.
Palavras chaves
MESP; orbital de fronteira; PM7
Introdução
O Tavaborole é um fármaco sintético antifúngico, indicado para o tratamento de onicomicose, devido a presença de Trichophyton rubrum ou Trichophyton mentagrophytes (HUI et al., 2007), esses fungos se alimentam da queratina, proteína que forma a maior parte das unhas, chegando a acometer até 14% da população, sendo mais frequente nos idosos (TREVISAN, 2016). Tavaborole possui um mecanismo de ação único, tornando-o altamente específico contra fungos patogênicos (MARKHAM, 2014). Ao contrário dos antifúngicos existentes, que inibem a síntese de ergosterol ou o metabolismo microbiano ,o tavaborole interfere com a síntese proteica em células fúngicas, possui um mecanismo de ação único, tornando-o altamente específico contra fungos patogênicos (MARKHAM, 2014). O objetivo desse trabalho foi utilizar rotinas de simulação computacional, configuradas para realizar cálculos quânticos semi-empíricos PM7 (Parametric Method 7) para caracterizar o fármaco sintético Tavaborole, estruturalmente e eletronicamente. Sendo o presente trabalho, uma etapa inicial para o melhoramento do fármaco, pois a partir do completo entendimento das características que influenciam a reatividade do composto, é possível iniciar o planejamento de novos compostos através de modificações estruturais.
Material e métodos
Para o desenvolvimento deste trabalho, foram utilizados softwares com licença gratuita para fins acadêmicos, baseados no Sistema Operacional Microsoft Windows®, utilizando um computador com processador intel® Core ™ i7-4510U, com 16,00 Gb de RAM com suporte de placa de vídeo AMD Radeon® com 2 Gb.No primeiro momento, foi realizada uma busca no repositório Drugbank (http://www.drugbank.ca/), utilizando o descritor TAVABOROLE, onde foram coletadas informações estruturais. No segundo momento, para realizar cálculos quânticos de abordagem semi-empírica, foi utilizada a metodologia proposta por Dewar e colaboradores (1985) para otimizar a estrutura e obter parâmetros energéticos (energia total, energia nuclear, energia eletrônica, energia dos orbitais moleculares de fronteiras, HOMO (HOMO-Highest Occupied Molecular Orbital) e LUMO (LUMO-Lowest Unoccupied Molecular Orbital), calor de formação). O software utilizado foi o programa Molecular Orbital Package (MOPAC2016), Versão 16.111W (STEWART, 2016), configurado para realizar método semi-empírico Parametric Method 7(PM7), usando a aproximação de Hartree-Fock (HF) (método de campo auto consistente), para função de onda, considerando a molécula no estado fundamental e no vácuo.
Resultado e discussão
A partir do repositório Drugbank, foi obtido propriedades básicas sobre o Tavaborole, destacando a sua estrutura bidimensional. Utilizando o método PM7, foi possível obter a estrutura conformacional de menor energia potencial (Fig. 1A). Obtivemos o mapa de superfície de potencial eletrostático (MESP), utilizado em estudos de relação estrutura/atividade, identificando os sítios reacionais da molécula, ou seja, os sítios polares (eletrofílicos e nucleofílicos) e regiões apolares da molécula (TASI et al., 1993). O potencial eletrostático é representado na forma de uma superfície de potencial eletrostático, onde uma maior densidade eletrônica é representada por cores quentes (vermelho, amarelo...) e uma menor densidade eletrônica por cores frias (azul, verde...) (ATKINS e PAULA, 2006). A região de maior densidade eletrônica, envolve o átomo de flúor (F1), gerando uma região com caraterísticas nucleofílicas e um sítio eletrofílico no átomo de hidrogênio (H12) (Fig. 1B). Baseados na teoria de Orbitais Moleculares de Fronteira (DOMINGO et al., 2003), cada molécula possui o seu orbital molecular ocupado de maior energia, HOMO e o orbital molecular desocupado de menor energia, LUMO, a energia de ambos os orbitais são utilizados para cálculos de descritores que caracterizam a reatividade da molécula. O HOMO, possui forma simétrica entra as fases positiva (azul) e negativa (vermelho) (Fig. 2A), é formado pela contribuição de todos os átomos de Carbono e Flúor, uma menor contribuição do átomo de Boro (B11). O LUMO (figura 2B), também apresenta uniformidade e simetria entre as fases positiva e negativa, formados pela contribuição de quase todos os átomos, com exceção dos átomos de Hidrogênio, ressaltando, que para o LUMO o átomo de Boro (B11) apresenta uma grande contribuição.
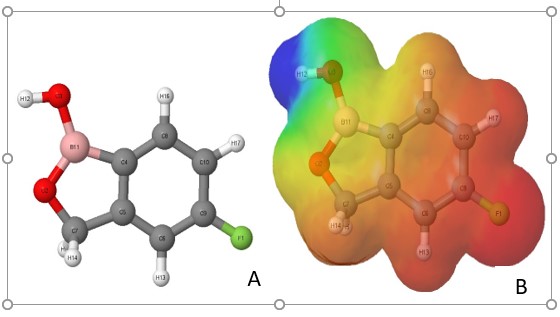
(A) Estrutura química da molécula do Tavaborole, otimizada utilizando o método semi-empírico PM7,(B) Mapa de superfície de potencial eletrostático do
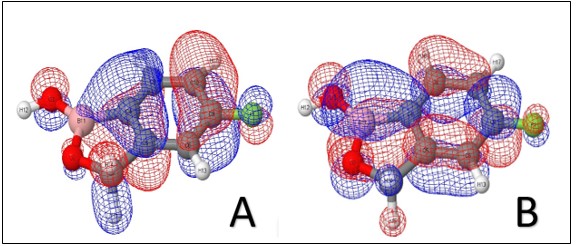
Orbitais moleculares de fronteira para o fármaco Tavaborole: HOMO (A) E LUMO (B).
Conclusões
A modelagem do fármaco Tavaborole, possibilitou obter sua estrutura conformacional de menor energia, identificar o átomo de Flúor como possível sítio reacional ( nucleofílico) , e um sítio eletrofílico no átomo de Hidrogênio (H12). Também foi possível plotar os orbitais moleculares de fronteira, que serão fundamentais para futuros cálculos de descritores moleculares, concluindo assim, que este trabalho constitui uma etapa inicial para o melhoramento do fármaco, e futuros estudos de modificações estruturais (Drug designer).
Agradecimentos
A Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes), e a Fundação Cearense de Apoio ao Desenvolvimento Cientifico e Tecnológico (FUNCAP) .
Referências
ATKINS, P.; de PAULA, J. Physical chemistry: Eighth Edition. Oxford University Press: Great Britain, 2006.
BARAK O, Loo DS. AN-2690, um novo antifúngico para o tratamento tópico da onicomicose. Curr Opin Investir Drogas. 2007; 8 (8): 662-668.
DOMINGO, L. R.; AURELL, M. J.; PÉREZ, P.; CONTRERAS, R.; J. Org. Chem.,v.68, p.3884, 2003.
ELEWSKI, B, E.; ALY, R.; BALDWIN, S, L.; SOTO, R, F, G.; RICH, P.; WEISFELD, M.; WILTZ, H.; ZANE, L, T.; POLLAK, R.; Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: Results from 2 randomized phase-III studies. Journal of the American academy of demartology, volume 73, issue 1, july 2015, pages 62-69.
FLICK, A. C., DING, H. X., LEVERETT, C. A., KYNE, R. E., LIU, K. K. C., FINK, S. J., & O’DONNELL, C. J. (2016). Synthetic approaches to the 2014 new drugs. Bioorganic & medicinal chemistry, 24(9), 1937-1980.
HUI, X., BAKER, S. J., WESTER, R. C., BARBADILLO, S., CASHMORE, A. K., SANDERS, V., & MAIBACH, H. I. (2007). In vitro penetration of a novel oxaborole antifungal (AN2690) into the human nail plate. Journal of pharmaceutical sciences, 96(10), 2622-2631.
LEWARS E.G. Computational chemistry: introduction to the theory of molecular and quantum mechanics. 2nd ed. Springe, 2011.
MARKHAM A. Tavaborole: primeira aprovação global. Drogas. 2014; 74 (13): 1555-1558.
ORTOLAN, A.O. Apostila de práticas de Química Computacional. 2014. 88p. Trabalho de conclusão do curso – Curso de Licenciatura em Química, Universidade Tecnológica Federal do Paraná. Pato Branco, 2014.
ROCK FL, Mao W, Yaremchuk A, et al. Um agente antifúngico inibe uma aminoacil-tRNA sintetase através da captura de ARNt no site de edição. Ciência. 2007; 316 (5832): 1759-1761.
STEWART, J. J. MOPAC2016, Version: 16.111W,.2016
TASI, G.; PALINKÓ, I.; NYERGES, L.; FEJES, P.; HORST, F. Calculation of electrostatic potential maps and atomic charges for large molecules. Journal of Chemical Information and Computational Science. n. 33, p. 296-299, 1993.
TREVISAN, F.; WERNER, B.; PINHEIRO, R.; Clipping ungeal na onicomicose e sua correlação com achados de cultura. Jornal Patologia Cutânea, CUP-O-382-2016.































