Síntese e Caracterização Estrutural de 1,2,4-Oxadiazóis Mediada por Energia de Ultrassom
ISBN 978-85-85905-25-5
Área
Iniciação Científica
Autores
França da Silva, A.R. (DQ/UFRPE) ; Luiz Moura, A. (DQ/UFRPE) ; de Freitas Filho, J.R. (DQ/UFRPE) ; de Freitas Filho, J.C. (CES/UFCG) ; Rufino de Freitas, J.J. (UACSA/UFRPE)
Resumo
Neste trabalho é descrito a síntese e caracterização estrutural de novos 1,2,4-oxadiazóis mediada por energia de ultrassom. Na primeira etapa reagimos diferentes arilnitrilas e cloridratos de hidroxilamina para fornecer as arilamidoxima (3a-h) (Ar= fenil, p-toluil, p-bromofenil, p-clorofenil, o- clorofenil, p-nitrofenil, 4-piridinil e piperonil). Em seguida, na segunda etapa, as arilamidoximas (3a-h) foi reagida com cianoacetato de etila para fornecer os novos 1,2,4-oxadiazóis (5a-h) em rendimentos que variam de 35% a 70%. Convém destacar que as arilamidoximas e os novos 1,2,4-oxadiazóis foram caracterizados por espectrometria de infravermelho e ressonância magnética nuclear de 1H e 13C.
Palavras chaves
Arilamidoxima; Oxadizóis; Ultrassom
Introdução
1,2,4-Oxadiazóis são uma classe de compostos quimicamente e biologicamente importante.1,2 Os 1,2,4-Oxadiazóis são conhecidos desde 1884 quando os pesquisadores alemães sintetizaram o primeiro composto desta série.3 As duas revisões das literaturas proposta por Clapp1,2 descrevem sobre as sínteses, a química, as reações, os estudos espectroscópicos e as atividades biológicas dos 1,2,4-oxadiazóis e 4,5-dihidro-1,2,4-oxadiazóis. Em 2014, Barros et al.4 obtiveram uma serie de 1,2,4 oxadiazóis partindo-se de arilamidoximas e do butanoato de etila usando irradiação de micro-ondas. Os autores testaram a atividade antitumoral in vitro dos compostos obtidos, onde três 1,2,4-oxadiazóis apresentaram atividades, pois bloquearam a proliferação das células tumorais.4 Atualmente, Os 1,2,4-oxadiazóis podem ser preparados por diferentes métodos, dentre eles destaque são dados para a) o aquecimento convencional, b) a irradiação de micro-ondas e c) energia de ultrassom. Baykov et al.,5 descreveram um método conveniente e suave para a síntese de 1,2,4-oxadiazol através de uma reação de ciclodehidratação de O-acilamidoximas em um sistema de superbase MOH / DMSO. Recentemente, Sağırlı e Dürüst (2018) descreveram a reação de 5- (clorometil)-3-substituído-fenil-1,2,4-oxadiazoles com KCN dando o tri- substituídos 1,2,4-oxadiazol-5-il-acetonitrilos e seus derivados.6 Neste trabalho descrevemos a síntese de uma nova série de 1,2,4-oxadiazóis (5a-h) mediada por energia de ultrassom.
Material e métodos
Nas etapas reacionais, utilizou-se reagentes e solventes na sua forma comercial, p.a. O acompanhamento das reações foi feito através de cromatografia em camada delgada (ccd). O solvente usado para correr a placa foi CH2Cl2/AcOEt (9:1). Para visualização dos compostos usamos lâmpada de ultravioleta e / ou cuba contendo vapores de iodo. Para cromatografia em coluna utilizamos sílica-gel 60 (Merck, 70 – 230 mesh). Os espectros de RMN 1H e 13C foram obtidos em espectrômetro VARIAN modelo Unity Plus (300 MHz), usando CDCl3 como solvente e tetrametilsilano como padrão interno. Espectros de infravermelho (IV) foram obtidos em espectrofotômetro de IV com Transformada de Fourier no instrumento BRUKER Modelo IFS 66. Inicialmente, realizou-se a preparação da arilamidoxima (Ar= fenil, p-toluil, p- bromofenil, p-clorofenil, o-clorofenil, p-nitrofenil, 4-piridinil e piperonil) utilizando o cloridrato de hidroxilamina e carbonato de potássio, em presença de etanol e água mediada por irradiação em ultrassom. A etapa seguinte consistiu em reagir, separadamente, as diferentes arilamidoximas com cianoacetato de etila em presença de hidróxido de sódio, 1 mL de DMSO utilizando a energia de ultrassom para produzir os novos 2-(3-aril-1,2,4- oxadiazol-5-il)-acetonitrila (5a-h). A segunda etapa reacional foi realizada usando ultrassom modelo UltraCleaner 1400A da Unique, de frequência 40KHz.
Resultado e discussão
Uma mistura de arilnitrilas (1a-h) e cloridrato de hidroxilamina 2 foi
convertida in situ nas correspondentes arilamidoximas (3a-h). Em seguida, as
arilamidoximas (Ar= fenil, p-toluil, p-bromofenil, p-clorofenil, o-
clorofenil, p-nitrofenil, 4-piridinil e piperonil) (3a-h) foram reagidas,
separadamente, com cianoacetato de etila (4) em presença de hidróxido de
sódio, 1 mL de DMSO e mediada sobre energia de ultrasom para
produzir os correspondentes 2-(3-aril-1,2,4-oxadiazol-5-il)-acetonitrila
(5a-h) (Esquema 1). Os novos 2-(3-aril-1,2,4-oxadiazol-5-il)-acetonitrila
(5a-h)) foram obtidos em tempos reacionais que variam de 5 - 7horas e com
rendimentos variando de 35% a 70%, como é mostrado na tabela 1. Também foram
realizadas
as medições dos pontos de fusão, os valores dos pontos de fusão estão na
tabela 1. As estruturas dos compostos sintetizados foram caracterizadas
pelas técnicas espectrométricas convencionais, IV, RMN1H e 13C. No espectro
de RMN de 1H do composto 5a pode-se observar um tripletos com integral para
3H, na região entre 8,49 ppm e 7,52 ppm referente aos hidrogênios H-3 e H-
4 do anel aromático, cuja constante de acoplamento foi de 6Hz. Na região
compreendida entre 8,06 ppm e 8,09 ppm aparecem um dupleto, com constante
de acoplamento de 6 Hz atribuídos ao hidrogênio 2 do anel aromáticos. O
próton metilênicos da cadeia lateral aparece na faixa de 4,10 ppm como um
simpleto. No espectro de RMN de 13C (Figura 2), pode-se observar um pico em
deslocamento 17,12 ppm referente ao carbono metilênico da molécula. Os
quatro (4) carbonos do anel aromáticos aparecem em deslocamentos 127, 129
e 131 ppm, enquanto carbonos dos anel 1,2,4-oxadiazol aparecem
característica na região de 168,29 ppm e 169 ppm .

Síntese de novos 1,2,4-oxadiazóis mediada por irradiação de micro-ondas.
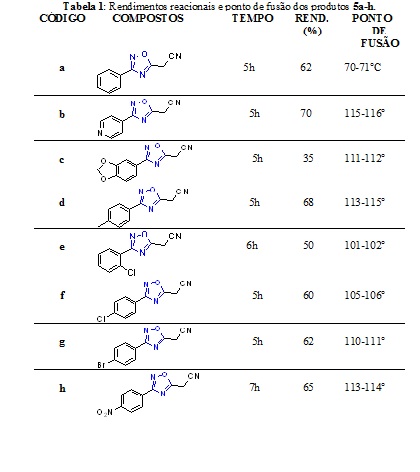
Rendimentos reacionais e ponto de fusão dos produtos 5a-h.

Espectro de RMN de 13C (75 MHz) em CDCl3 do composto a

Espectro de RMN de 13C (75 MHz) em CDCl3 do composto a.
Conclusões
Em resumo, descrevemos a síntese e novos 2-(3-aril-1,2,4-oxadiazol-5-il)- acetonitrila (5a-h). Os produtos finais foram obtidos em tempos que variam de 5 - 7horas e com rendimentos moderados (35-75%). As estruturas dos compostos foram elucidadas através da espectrometria de infravermelho ( IV) , RMN (1H e 13C) como também analise elementar.
Agradecimentos
FACEPE, CAPES e CNPQ pelo suporte financeiro
Referências
1 TIEMANN, F; KRÜGER, P. Ber. 17, 1685, 1984.
2CLAPP, L. B. in Advance in Heterocyclic Chemistry , Katritzky, A. R.; Ed., Academic Press, New York, N.Y., 20, 65, 1976.
3CLAPP, L. B. in Comprehensive Heterocyclic Chemistry, Katritzky A. R. and Rees, C. W. (Eds.), Pergaman Press, 6, 365, 1984 and references cited therein.
4BARROS, C. J. P.; SOUZA, Z. C.; FREITAS, J. J. R.; DA SILVA, P. B. N.; MILITÃO, G. C. G.; SILVA, T. G.; FREITAS, J. C. R.; FILHO, J. R. F. J. Chil. Chem. Soc., 59, nº 1, 2359-2362. 2014.
5BAYKOV, S.; SHARONOVA, T.; OSIPYAN, A.; ROZHKOV, S.; SHETNEV, A.; SMIRNOV, A.. Tetrahedron Letters., 57, 2898, 2016.








