TÍTULO: Modelagem por homologia de receptores canabinóides CB1 e CB2 e estudos de docking
AUTORES: da Silva, J.A. (UNIVERSIDADE FEDERAL DE ALAGOAS) ; Silva, T.M.B. () ; Balliano, T.L. ()
RESUMO:Avaliou se os dados obtidos com os disponíveis na literatura, de moléculas como
potenciais inibidores para receptores canabinóides CB1 e CB2, onde foi feito um
estudo de modelagem molecular com CB1 e CB2, cuja estrutura tridimensional não
disponível em banco de dados, foi obtida por modelagem por homologia; seguido de
uma simulação de docking para o qual usamos moléculas de estrutura do sistema
monoterpeno
PALAVRAS CHAVES: Modelagem por homologia; receptores canabinóides; docking
INTRODUÇÃO:No planejamento de um fármaco tem como base a informação estrutural do bioreceptor
que permite a descoberta e síntese de compostos com complementaridade estérica,
hidrofóbica e eletrostática ao seu sítio de ligação. Em todo este processo usamos
a modelagem molecular como ferramenta. Como para os receptores canabinóides CB1 e
CB2, o alvo protéico macromolecular ainda não foi determinado experimentalmente,
construímos os modelos em um procedimento comparativo conhecido como modelagem
molecular por homologia estrutural (SANTOS-FILHO; DEALENCASTRO,2003). Os
canabinóides são uma classe de compostos que exerçam efeitos farmacológicos
diretos sobre um determinado número de órgãos e normalmente relacionados ao
sistema nervoso central.
MATERIAL E MÉTODOS:Para a obtenção dos modelos das proteínas CB1 e CB2 foi usado arquivos FASTA
obtidos do banco de dados NCBI, então foi usado o software Modeller (SALI;
BLUNDELL, 1993). Em seguida foi feito a simulação de docking como programa
Autodock 4.2 (que usa algoritmo de busca, o algoritmo genético Lamarkiano), com um
conjunto de moléculas das quais os melhores scores foi obtido com o ligante 1: 1-
Hidroxi-6,6-dimetil-3-pentil-6H-benzo[c]cromeno-9-ácido carboxílico e ligante 2:
6,6,9-Trimetil-3-pentil-6H –benzo[c]cromeno-1,8-diol. O ancoramento foi verificado
no sítio de ligação previsto por LIGSITE (HENDLICH; RIPPMANN, 1997). Desta forma
obtemos dados de energia de ligação estimada, coeficiente de inibição e modo de
ligação que puderam ser avaliados e comparados dados de referência (HOWLETt 2002).
RESULTADOS E DISCUSSÃO:Obtemos modelos C1 e CB2 com o programa Modeller e verificado a qualidade do
modelo com o programa PROCHECK, que mostra que apenas poucos resíduos (1,8 %
CB1 e 0,6 % CB2) estão em regiões desfavoráveis para o gráfico Ramachandran. Com
a simulação de docking obtemos para CB1 Ligante 1, energia de ligação E= -9.09
kcal/mol e coeficiente de inibição Ki = 217.43 nM (nanomolar), interagindo com
os resíduos Ser88 (ligação de hidrogênio de 1,82 A),Gly99, Trp241, Phe191; o
Ligante 2 com a proteína CB2, E= -9.79 kcal/mol e Ki = 66.40 nM (nanomolar)
interagindo com os resíduos tyr347, Leu76, Glu327 (1,97 A) e Lys219 (1,99 A);
nos dois caso podemos verificar a possibilidade de interação com os resíduos do
sítio de ligação conforme previsto pelo programa LIGSITE (Leis, Schneider ;
Zacharias, 2010), os quais podem ser visto nas figuras 1 e 2 em anexo.
Forma prevista do modo de ligação do Ligante 1, no sítio de ligação da proteína
CB1.
Figura1
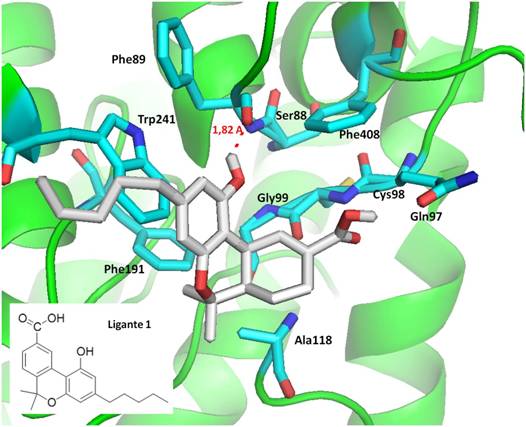
Forma prevista do modo de ligação do Ligante 1, no
sítio de ligação da proteína CB1.
Figura 2
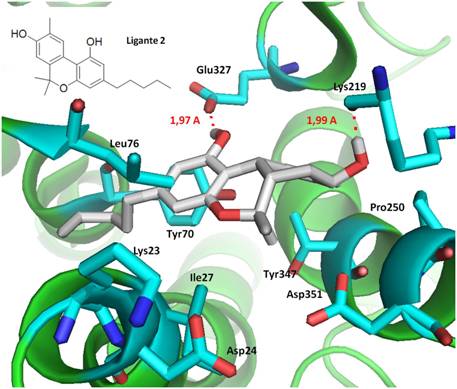
Forma prevista do modo de ligação do Ligante 2, no
sítio de ligação da proteína CB2.
CONCLUSÕES:Segundo os dados da literatura (HOWLWETT 2002), (MONTERO, et al, 2010) os Ligante
1 e 2 tem as melhores potencialidades dentro do conjunto de moléculas testadas,
com relação aos valores de coeficiente de inibição estimados e com as
características relacionadas a seletividade em relação aos
receptores,característica desejável (DA SILVA; SILVA, 2006) visto que os ligantes
1 e 2 deve ter afinidade por um receptor canabinóide e pouca pelo outro. Assim o
Ligante 1 teve E= -8.16 kcal/mol e Ki = 1.05 uM (micromolar); já para o Ligante 2
com CB, tivemos E= -8.18 kcal/mol e Ki = 1.01 uM (micromolar).
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA:DA SILVA, .V. ; SILVA H.T.P Revista eletrônica de farmácia Vol. IV (1), 15-26, 2007
SANTOS-FILHO, O. A. DEALENCASTRO, R. B Quim. Nova, Vol. 26, No. 2, 253-259, 2003
SIMON LEIS, SEBASTIAN SCHENIDER AND MARTIN ZACHARIAS In Silico Prediction of Binding Sites on Proteins;. Current Medicinal Chemistry, 2010, 17, 1550-1562
MONTEIRO, C; CAMPILLO,N. E.; GOYA. European Journal of Medicinal Chemistry 40 (2005) 75–83
SALI, A.; BLUNDELL, T. L. Comparative protein modeling by satisfaction of spatial restraints. Journal of Molecular Biology. v. 234, p. 779-815, 1993.
HOWLETT et al (2002) Pharmacol.Rev. 54 161.
