
ÁREA: Físico-Química
TÍTULO: ESTUDO DA ESTRUTURA, ISOMERIA CONFORMACIONAL E CENTROS ATIVOS DO ÁCIDO ISOBRASILIÊNSICO
AUTORES: SILVA, S. C. DA - UFMT (PQ)
TEIXEIRA, D. A. - UFMT (PG)
CERON, L. B. - UFMT (IC)
RESUMO: O trabalho desenvolvido através de cálculos quânticos computacionais gerou vários isômeros conformacionais do ácido isobrasiliênsico, que apresentaram uma pequena diferença de energia. A molécula possui atividade antiúlcera, e procurou-se estudar o seu comportamento em meio ácido para encontrar os possíveis centros reacionais da molécula. Foram efetuados cálculos quânticos ab initio (HF e DFT) e semi-empíricos (AM1 e PM3) para as otimizações das estruturas dos isômeros conformacionais, para a obtenção dos orbitais moleculares e reações de protonação. Foram encontrados vários pontos de protonação energeticamente favoráveis. Com a reação de protonação obteve-se várias estruturas estáveis, entre as quais três produtos de reação de ciclização apresentaram energias mais baixas.
PALAVRAS CHAVES: ácido isobrasiliênsico; modelagem molecular; química computacional.
INTRODUÇÃO: O progresso da tecnologia de hardware e software, na últimas décadas do século XX, tem permitido um grande desenvolvimento da química computacional, tornando-a uma das áreas mais promissoras deste início de século XXI.
O composto éster metilico do ácido 6-O-metil isobrasiliênsico (C32H46O6) foi isolado e caracterizado no Laboratório de Produtos Naturais UFMT (Caneppele,1998). Este composto foi extraído da casca do caule de Calophyllum brasiliense CAMB, conhecida por apresentar atividade antiúlcera.
Através da dinâmica molecular com aquecimento controlado gerou-se isômeros conformacionais do ácido isobrasiliênsico. Cada um dos isômeros gerados foi otimizado com base em cálculos quânticos, utilizando métodos semi-empíricos e ab initio. A partir da análise de orbitais moleculares foram identificados possíveis centros reativos, e foi feito um estudo de reações de protonação nestes sítios.
Com o estudo de propriedades físico-químicas utilizando a modelagem molecular, espera-se contribuir para um melhor conhecimento da estrutura e propriedades desta molécula, bem como de sua reatividade e seus mecanismos de reação em meio ácido.
MATERIAL E MÉTODOS: O trabalho foi realizado em um computador Pentium IV, 3.0 GigaHertz, com 1GByts de memória RAM. A molécula foi construída com o programa HyperChem. Os isômeros conformacionais foram obtidos com o mesmo programa, pelo método da dinâmica molecular através de aquecimento simulado, o qual permite a variação de todas as distâncias e ângulos de ligação da molécula. Foram gerados 11 isômeros conformacionais. Otimizou-se as geometrias de cada um dos isômeros com a utilização do programa GAUSSIAN 03W. Numa primeira etapa foi feita a otimização utilizando-se os métodos semi-empíricos PM3 e AM1. Em seguida foram feitas otimizações utilizando métodos ab initio, HF com a base LANL2DZ e o método DFT junto com a base B3LYP/6-31G(YOUNG, C. D. 2001).
Com o objetivo de encontrar possíveis centros doadores e receptores de elétrons calculou-se os orbitais moleculares de fronteira HOMO, HOMO-1, HOMO-2, LUMO, LUMO+1, LUMO+2, para cada isômero conformacional (JENSEN, F. 1999). Para estes cálculos foi utilizado o método HF/LANL2DZ.
Finalmente, com base nos cálculos de orbitais moleculares, simulou-se reações de protonação em vários pontos da molécula, utilizando o método semi-empírico PM3.
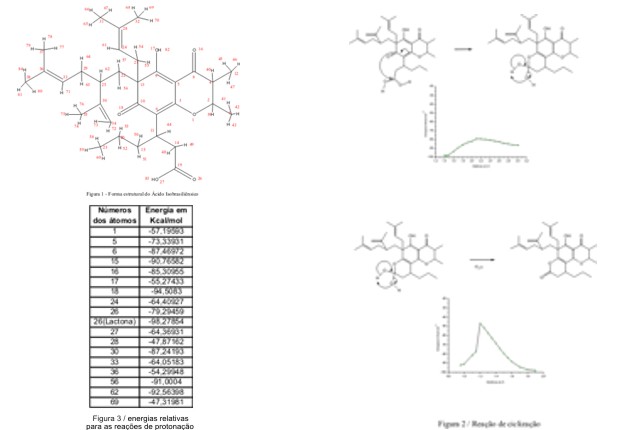
RESULTADOS E DISCUSSÃO: O ácido isobrasiliênsico (figura 1) possui grupos funcionais como, ácido carboxílico, álcool, cetona e éter e apresenta ramificações que permitem a existência de grande quantidade de isômeros conformacionais. Os cálculos quânticos efetuados mostraram que estes isômeros apresentaram energias semelhantes. Isto sugere que no equilíbrio à temperatura ambiente, o sistema apresenta uma mistura de vários isômeros em quantidades equivalentes.
Com as reações de protonação obteve-se vários produtos estáveis e com energia de ativação bastante baixa, sugerindo que em meio ácido a molécula se decompõe espontaneamente. Na figura 2. é mostrado que a protonação de um grupo carboxílico leva a uma reação de ciclização e posterior liberação de água. Na primeira etapa desta reação a energia de ativação de 7,20 Kcal.mol-1, e na segunda é da ordem de 45 Kcal.mol-1. Na figura 3 são mostradas as energias relativas para as reações de protonação em diferentes átomos da molécula, numerados conforme a figura 1.
Os cálculos das densidades eletrônicas dos orbitais HOMO, HOMO-1, HOMO-2, LUMO, LUMO+1, LUMO+2, confirmaram as possibilidades de protonação e os rearranjos na estrutura molecular.
CONCLUSÕES: O ácido isobrasiliênsico apresenta três ramificações, o que permite a formação de vários isômeros conformacionais. Os valores de energia para cada isômero foram semelhantes, sugerindo que o ácido isobrasiliênsico se apresente em várias conformações em temperatura ambiente. A simulação de reação de protonação com base nos cálculos dos orbitais moleculares de fronteira, sugere a possibilidade de formação de vários produtos de decomposição do ácido isobrasiliênsico, exigindo assim, estudos posteriores para se identificar o composto que apresenta efetivamente atividade farmacológica.
AGRADECIMENTOS:CNPq / FAPEMAT
REFERÊNCIAS BIBLIOGRÁFICA:CANEPPELE, Décio Estudo Químico De Constituintes Com Potencial Atividade Antiúlcera Da Casca do Caule De Calophyllm brasiliense CAMB. (GUANANDI). 1998. 134p. Dissertação (Mestrado em saúde e ambiente) Instituto de Saúde Coletiva, Universidade Federal de Mato Grosso, Cuiabá, MT, 1998.
YOUNG, C. David. Computational Chemistry. WILEY-INTERSCIENCE (USA), 2001, 371p.
JENSEN, F. Introduction to Computational Chemistry. John Wiley & Sons, New York (USA), 1999.