ÁREA: Físico-Química
TÍTULO: DESENVOLVIMENTO DE FÁRMACOS DERIVADOS DA DIIDROARTEMISININA CONTRA MALÁRIA USANDO MÉTODOS DE QUÍMICA QUÂNTICA E QUIMIOMÉTRICOS
AUTORES: SANTOS, C.B.R (UEAP) ; GOMES, T.L.D (UFPA) ; MACEDO, W. J.C (UFPA) ; MACIEL, A. A. (UFPA) ; PINHEIRO, J. C (UFPA)
RESUMO: Derivados da diidroartemisinina com atividade antimalárica, resistente à cloroquina e sensível à mefloquina, são propostos usando-se métodos de química quântica e quimiométricos. A aplicação de PCA e HCA mostrou que os descritores responsáveis pela classificação dos compostos em mais ativos e menos ativos foram à energia de hidratação (EH), carga sobre o átomo de oxigênio O11 (QO11), ângulo de torção O2-O1-Fe-N2 (D2) e o índice máximo de R/Eletronegatividade de Sanderson (RTe+). Os mapas de potencial eletrostático molecular foram computados pelas cargas de Mullikan com o conjunto de base de valência HF/6-31G** e comparados qualitativamente na região do anel trioxano da artemisinina para predizer se os novos derivados apresentam atividade antimalarial.
PALAVRAS CHAVES: diidroartemisinina, pca/hca e mep
INTRODUÇÃO: Um grande número de drogas tem sido investigado no tratamento da malária, entretanto, novas cepas de plasmodium falciparum resistentes a algumas dessas drogas tem causado deteriorização no tratamento clinico dessa doença. A artemisinina é um extrato químico obtido da Artemísia annua, pouco solúvel em água, que tem sido eficaz no tratamento do ataque agudo da malaria tanto do plasmodium vivax quanto do plasmodium falciparum (KLAYMAN, 1985).
Na atualidade o desenvolvimento racional de fármacos consiste na modificação molecular considerando um composto de estrutura química conhecida e ação biológica comprovada como modelo para ensaio de novos compostos que sejam análogos estruturais do fármaco matriz. Este método quando utilizado requer elevado tempo e alto investimento financeiro na obtenção de um novo fármaco.
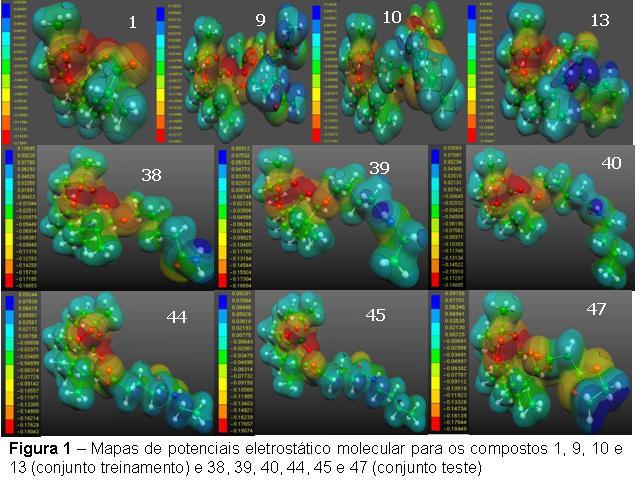
Neste trabalho, usamos métodos de química quântica e quimiométricos para os novos derivados da diidroartemisinina. Os mapas de potencial eletrostático molecular (MEP's) foram avaliados e usados para identificar características chaves responsáveis pela atividade destes novos derivados. De acordo com a literatura a forma geométrica do potencial eletrostático da artemisinina quando comparada com os seus derivados, apresentam mapas de potencial eletrostático molecular com forma similar àquela da artemisinina na região do anel 1,2,4-trioxano para todos os compostos ativos (BERNARDINEILI et al, 1994).
MATERIAL E MÉTODOS: As estruturas dos compostos do conjunto de treinamento e do conjunto teste foram construídos com o programa Gauss View 1.0, de acordo com o seguinte procedimento: inicialmente a estrutura da artemisinina foi construída e otimizada com o método ab initio HF/6-31G**, e em seguida, os demais compostos do conjunto treinamento (1-21) e conjunto teste (22-51) foram construídos a partir da geometria da artemisinina, e dessa forma obteve-se as estruturas otimizadas com menor energia, sendo o conjunto treinamento (1-21) testados in vitro em sangue humano parasitado por cepas de malária W-2 oriunda da Indochina, causada por plasmodium falciparum (AVERY et al, 1993).
A qualidade do conjunto de base de valência foi verificada comparando os parâmetros geométricos calculados do anel 1,2,4-trioxano com os resultados experimentais (PINHEIRO et al, 2001). A conformação mais estável de cada molécula foi usada para realização dos cálculos dos descritores:
(a) Descritores Químico-Quânticos e QSAR, foram calculados usando o programa Gaussian 98 e HyperChem 6.02, respectivamente;
(b) Descritores GETWAY, foram calculados usando o programa Dragon 5.4;
(c) Descritores Docking Molecular, foram realizados usando programa AutoDock 2.4.
O potencial eletrostático molecular foi obtido através de um conjunto de cargas pontuais de modo que representasse o potencial quântico molecular melhor possível de pontos definidos em torno da molécula. Os mapas do potencial eletrostático molecular representa uma superfície de contorno, e estes foram computados pelas cargas de Millikan e pelas palavras chaves GFPRINT POP=FULL.
RESULTADOS E DISCUSSÃO: Na Figura 1, são mostrados os mapas de potencial eletrostático molecular da artemisinina e de seus derivados para as moléculas estudadas, onde quatro moléculas (conjunto de treinamento) e seis moléculas (conjunto teste), tem forma similar na região do anel 1,2,4-trioxano da artemisinina (1).
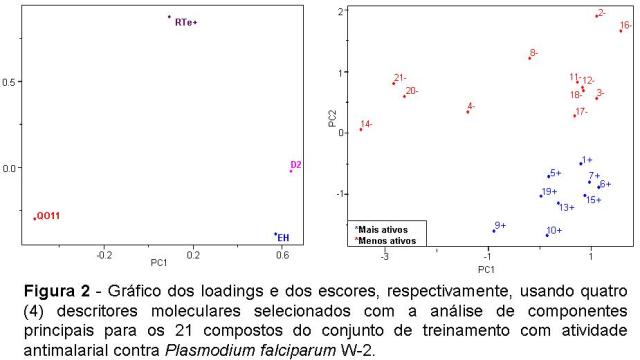
A análise PCA e HCA dos dados facilitou a seleção de 4 descritores (EH, QO11, D2 e RTe+) responsáveis pela classificação dos compostos em mais ativos e menos ativos. Antes de aplicarmos os métodos cada uma das variáveis foi auto escalada para podermos compará-las na mesma escala. A melhor separação foi obtida em PC2 que discrimina os compostos em mais ativo e menos ativo. Isso sugeriu que os demais descritores não são significativos na classificação desses compostos. Os resultados do PCA mostraram que os 3 primeiros PCs (PC1, PC2 e PC3) descrevem 93,9461% da informação total, perdendo apenas 6,0539% da informação original. Podemos verificar na Figura 2 que os descritores QO11, D2 e EH são os responsáveis pelo deslocamento dos compostos mais ativos, enquanto que o descritor RTe+ desloca os compostos menos.
Os resultados obtidos pela análise hierarquia de closter (HCA) foram similares àqueles obtidos pelo PCA.
CONCLUSÕES: Artemisinina e seus derivados foram estudados com o uso de métodos de química quântica e quimiométricos.
A aplicação de PCA e HCA permitiu a separação dos compostos do conjunto de treinamento em mais ativos e menos ativos e os descritores responsáveis pela classificação dos compostos foram à energia de hidratação (EH), carga sobre o átomo de oxigênio O11 (QO11), ângulo de torção O2-O1-Fe-N2 (D2) e o índice máximo de R/Eletronegatividade de Sanderson (RTe+).
O estudo dos MEP's mostrou que os compostos mais ativos do conjunto teste tem MEP similar aqueles dos compostos do conjunto treinamento.
AGRADECIMENTOS: UEAP - UNIVERSIDADE DO ESTADO DO AMAPÁ;
UFPA/LQTC - UNIVERSIDADE FEDERAL DO PARÁ/LABORATÓRIO DE QUÍMICA TEÓRICA COMPUTACIONAL.
REFERÊNCIAS BIBLIOGRÁFICA: KLAYMAN, D. L. Science, 1985, 228, 1049-1055.
BERNADINELLI et al. International Journal of Quantum Chemistry: Quantum Biology Symposium, 1994, 21, 117-131.
AVERY, M. A. Journal of Medicinal Chemistry, 1993, 36, 4264-4275.
PINHEIRO, J. C.; FERREIRA, M. M. C.; ROMERO, O. A. S. Journal Molecular Structure (THEOCHEM), 2001, 572, 35-44.
