ÁREA: Química Tecnológica
TÍTULO: INVESTIGAÇÃO COMPUTACIONAL DE ISÔMEROS DO AMINOÁCIDO GLICINA
AUTORES: BARROS, H.C. (UFPA) ; BRITO, H.G. (UFPA) ; SILVA, J.P. (UFPA) ; DAMSCENO, T.S (UFPA) ; OLIVEIRA, P.M.M (UFPA) ; CAMPOS, W.E.O (UFPA)
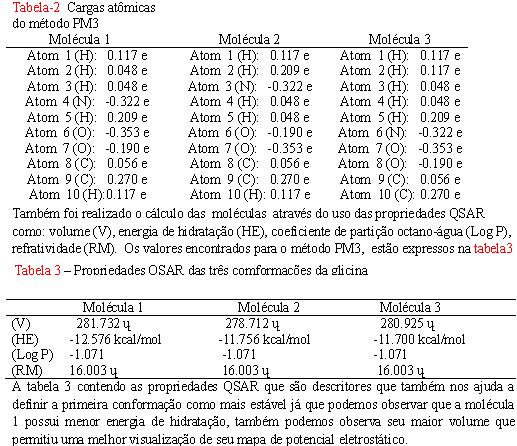
RESUMO: Esse trabalho tem como objetivo investigar nos isômeros da glicina propriedades físicas e químicas com o auxílio de cálculos computacionais podendo obter vários parâmetros como: volume (V), energia de hidratação (HE), coeficiente de partição octano-água (Log P), refratividade (RM) , momento dipolo (µ) e interpretar mapa de potencial eletrostático, esses cálculos foram realizado pelo método semi-empirico PM3, que tem apresentado resultados mais próximos aos obtido experimentalmente e por cálculos ab initio e comprovar que a modelagem molecular é muito importante para determinar atividades de moléculas na farmacologia reduzindo tempo e gastos.
PALAVRAS CHAVES: investigação ,computacional, glicina
INTRODUÇÃO:
A química computacional na atualidade é uma ferramenta cada vez mais útil e desejável para o ensino e pesquisa, para estudante tanto do ensino médio quanto superior. No ensino da química é possível obter resultados altamente confiáveis de cálculos de propriedades, sendo aplicada com sucesso no estudo de uma ampla faixa de problemas de interesse químico, como estabilidade conformacional e se pode calcular com maior precisão os parâmetros atômicos.
A precisão dos cálculos computacionais é impressionante mesmo para estruturas complexas, o desvio entre os valores calculados e os valores medidos experimentalmente é menor que 10%,dando um enorme grau de acerto a uma estrutura microscópica, composta por um sistema de muitos corpos (vários elétrons e o núcleo atômico).[1]
A proposta é estudar três isômeros da molécula glicina que é um dos aminoácidos codificados pelo código genético, sendo, portanto um dos componentes das proteínas dos seres vivos. Devido à sua simplicidade estrutural, este aminoácido tende a ser conservado evolucionariamente em proteínas como o citocromo, a mioglobina e a hemoglobina. A glicina é um aminoácido que apresenta um alto grau de flexibilidade quando integrado numa cadeia polipeptídica, Cada isômero é um mínimo local sobre a superfície da energia (o chamado potencial energético superfície), criado a partir do total de energia (ou seja, a energia eletrônica, Plus a repulsa energia entre os núcleos).
MATERIAL E MÉTODOS: No desenvolvimento dos métodos computacionais, surgiram os métodos de ajustes semi-empíricos. Estes códigos numéricos foram flexibilizados de maneira que alguns parâmetros energéticos pudessem ser ajustados e outros fossem mantidos fixos e iguais aos valores experimentalmente conhecidos. No final, um cálculo parcialmente teórico e parcialmente experimental pode ser realizado. Além de melhorar a precisão computacional e assim melhor orientar o trabalho experimental na procura dos valores para novos parâmetros atômicos. No método semi-empirico PM3, os núcleos são assumidos em sucessivas posições estacionárias, sobre as quais a distribuição espacial ótima dos elétrons é calculada pela resolução da equaçãode Schrödinger Eq.[1]. O processo repetido até se alcançar um ponto estacionário da superfície de energia. Em um sistema no estado fundamental, isto significa que a geometria é tal que o calor de formação (∆Hf) é um mínimo irredutível, ou seja, todas as suas constantes de força são positivas; para estados de transição, o sistema deve ter exatamente uma constante de força negativa. Deste modo, tem-se tornado prática comum nos trabalhos teóricos de qualidade a avaliação de todas as segundas derivadas (constantes de força) da energia molecular em função dos parâmetros moleculares, para se determinar inequivocamente a natureza dos pontos estacionários encontrados no processo de otimização da geometria molecular. O programa hyperchem 7.5 também foi utilizado para visualização do potencial eletrostático molecular, que representa uma superfície de contato obtida a partir das cargas de Mullikan, sendo então possível realizar movimentos tridimensionais das estruturas, possibilitam a observação de uma estrutura microscópica através de um modelo macroscópico
RESULTADOS E DISCUSSÃO: Para os três isômeros das moléculas glicina o semi-empirico PM3 foi o método escolhido e mais adequado, pois teoricamente é o que melhor descreve as propriedades estruturais e eletrônicas dos compostos estudados. Os valores dos calores de formação (∆Hf) e a construção das estruturas possibilitou o melhor entendimento dos isômeros. Para o primeiro isômero fig.[1b] obteve-se ∆Hf em (-2454,79492 Kcal/mol), que é 4,16217 Kcal/mol mais estável que o segundo isômero fig.[2b] e é 1,1249 mais estável que o terceiro isômero fig.[3b]. Isto também é comprovado pelo momento dipolo de cada estrutura I- (µ = 1,032 D) ; II- (µ = 5,016 D) ; III- (µ = 1,417).
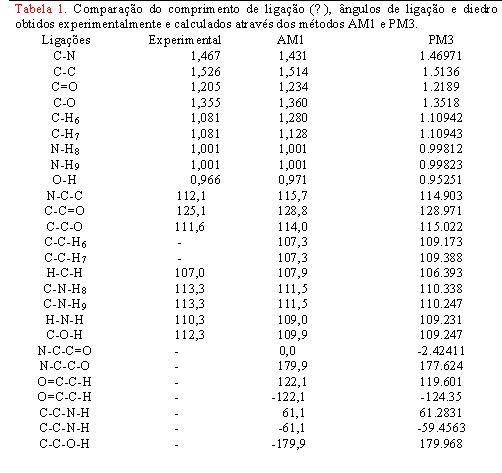
Devido sua maior estabilidade fizemos uma comparação utilizando valores de comprimento da ligação, ângulo de ligação e ângulo diedro do primeiro isômero otimizado usando o método PM3, com os valores experimentais e valores do método AM1 encontrados na literatura, comprovando que o método PM3 é muito eficaz pois seus valores estão bem próximo do experimental Tab.[1]
CONCLUSÕES: Pela análise dos resultados apresentados concluímos que o uso de modelagem computacional possibilita abordar um número maior de tópicos e um aprofundamento da teoria. E que o método semi-empírico PM3 descreveu bem os resultados comparando com o experimental.
A investigação computacional dos calores de formação foi possível predizer que a conformação 1 da glicina é a mais estável.
E evidenciar as propriedades QSAR e os mapas de potencial eletrostático são suportes de grande importância no estudo de moléculas como os aminoácidos que apresentam importância conformacionais na farmacologia.
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA: [1] - http://www.if.uff.br/plasma/portugues.htm
[2] - Copyright 2002 Hypercube, Inc.
[3] - Plukiger, P.; Lutthi, H. P.; Portmann, S.; Webber, J.; MOLEKEL 4.1, Swiiss Center for Scientific Computing, Switzerland, 2000-2001
[4] - Pinheiro, J. C., Ferreira, M.M.C., Romero, O.A.S., Antimalarial activity of dihydroartemisinin derivatives against P. falciparum resistent to mefloquine: a quantum chemical and multivariate satudy. Journal of Molecular Structure (Theochem) 572 (2001) 35-44.
[5] - Cardoso, F.J.B., Figueiredo, A.F., Lobato, M.S., Miranda, R.M., Almeida, R.C.O.A., A Study on Antimalarial Artemisinin derivatives using MEP maps and Multivariate QSAR. Journal of Molecular Modeling, aceito 2007.
