ÁREA: Físico-Química
TÍTULO: Estudo Teórico do mecanismo de formação de alilamidas a partir de adutos de Morita-Baylis-Hillman.
AUTORES: ZARAMELLO, L. (UFSC) ; CARAMORI, G.F. (UFSC) ; FERREIRA, M. (UFSC) ; FAGGION, D.J. (UFSC) ; SA, M.M. (UFSC) ; DOMINGOS, J.B. (UFSC)
RESUMO: O mecanismo para a formação das alilacetamidas 5a e 5b a partir do alfa-metileno-beta-hidroxi éster 1 (aduto de Morita-Baylis-Hillman, MBH) foi estudado por meio de cálculos DFT. Estudaram-se três possíveis mecanismos, SN1, SN1 e SN2. Os cálculos mostram que há preferência pelo mecanismo SN1 com formação de um carbocátion no estado de transição. No entanto, as barreiras energéticas para os mecanismos SN1 e SN1 são muito próximas ocorrendo uma competição entre estes dois mecanismos, o que está de acordo com resultados obtidos experimentalmente de Relação Linear de Energia Livre de Hammett que indicam que há forte formação de uma carga parcial positiva no estado de transição da etapa determinante.(FAGGION et al., 2009)
PALAVRAS CHAVES: cálculos dft, alilamidas, morita-baylis-hillman.
INTRODUÇÃO: Reações utilizando adutos de Morita-Baylis-Hillman (MBH) como intermediários sintéticos são amplamente empregadas em síntese orgânica na preparação de novos compostos. (SINGH et al., 2008). Estes adutos podem ser convertidos em uma série de compostos que apresentam excelente régio e estereosseletividade, além de requerer condições brandas de reação (COELHO et al., 2000). Alilamidas também são importantes intermediários na síntese de fármacos (MONK et al., 1991), podendo ser obtidas a partir de reação de um aduto de MBH com acetonitrila catalisada por ácidos. (FERREIRA et al., 2009). Assim, estudos teóricos através de cálculos DFT foram realizados para complementar os dados experimentais de Relação Linear de Energia Livre de Hammett (CAREY et al., 2007) se propor um mecanismo para esta reação.
O mecanismo para a formação de alilamidas 5a e 5b a partir de adutos de MBH 1 foi estudado através de cálculos utilizando a Teoria Funcional da Densidade (DFT), a fim de investigar por qual tipo de substituição nucleofílica a reação procede. Avaliamos se o mecanismo é do tipo SN1 ou SN1 por passos, com formação de um intermediário carbocátion 3, ou um mecanismo concertado do tipo SN2, com ataque da acetonitrila ao carbono alílico de 2 e deslocalização de elétrons para migração da dupla ligação e saída de água formando o cátion nitrílio 4a (Figura 1). Este segundo mecanismo difere do SN2 apenas no sítio de ataque do nucleófilo, neste o ataque ocorre no carbono ligado à hidroxila protonada. (BORDWELL et al., 1970).
MATERIAL E MÉTODOS: Todos os cálculos foram efetuados usando o programa Gaussian03 (POPLE, et al.,2003) no cluster Minerva do Departamento de Física da USFC.
As geometrias de mínimo e TS foram otimizadas com conjunto de funções de base B3LYP/TZVP. Os cálculos foram realizados com geometrias no vácuo (estado gasoso) onde não são considerados nenhum efeito de solvatação. As geometrias de mínimo e estados de transição (TS) foram otimizadas empregando-se o modelo B3LYP/TZVP. As estruturas dos estados de transição foram determinadas a partir da análise da matriz hessiana. Foram efetuados também cálculos da coordenada intrínseca de reação (IRC) a fim de comprovar se a geometria encontrada como TS conectava dois mínimos de interesse na superfície de energia.
RESULTADOS E DISCUSSÃO: A figura 1 ilustra os três mecanismos estudados. O foco do estudo foi a formação dos cátions nitrílio 4a e 4b, a formação das alilamidas 5a e 5b ocorre após hidrólise destes cátions.
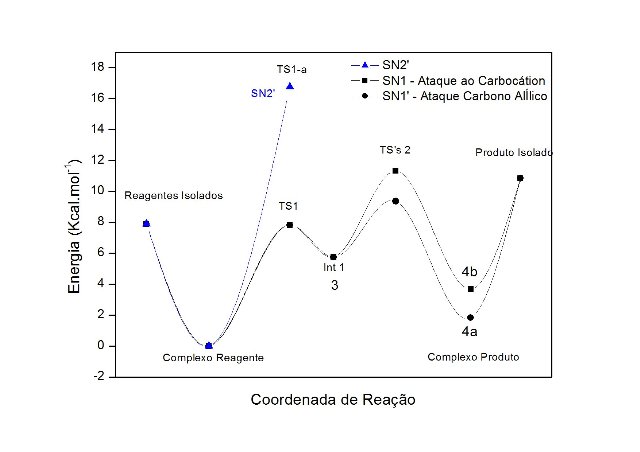
A partir da geometria protonada, avaliou-se as diferenças nas barreiras energéticas para os mecanismos do tipo SN1, SN1 e SN2. Os valores das barreiras são mostradas no Gráfico (figura 2).
A barreira para formação do intermediário carbocátion após a saída da água é 7,82 kcal/mol, seguida pelo ataque da acetonitrila no carbono benzílico (SN1) ou no carbono alílico (SN1), com barreiras energéticas de 5,58 e 3,62 kcal/mol, respectivamente. A barreira energética total para formação dos cátions nitrílio 4a e 4b é 9,36 (SN1) e de 11,35 kcal/mol (SN1).
Como se observa no gráfico, o mecanismo do tipo SN2 possui uma barreira de 18,16 kcal.mol-1 e mesmo após exaustivas buscas não foi observado um único estado de transição para a saída da água simultaneamente ao ataque da acetonitrila ao carbono alílico isto sugere que a reação procede através de um mecanismo por passos. Estes resultados são reforçados por estudos, ainda não publicados, da cinética desta reação, onde os dados obtidos através da Relação Linear de Energia Livre de Hammett mostram que há uma melhor correlação com a constante do substituinte sigma+ (R=O,9916), do que com sigma (R=O,6672), apresentando uma constante de reação (ro) de -4,39, o que sugere uma forte formação de carga positiva (carbocátion) no estado de transição e a ressonância direta desta com o anel aromático. A análise do valor de energia para o complexo produto SN1 mostra que a geometria do produto final além de ser o produto cinético, também é produto termodinâmico da reação (4a).
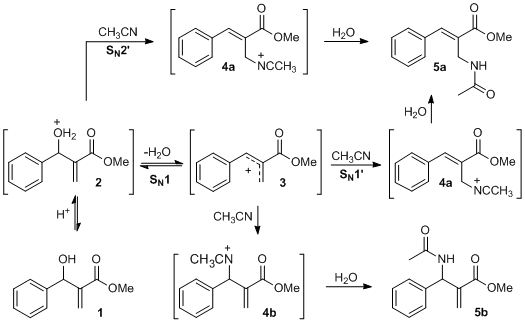
CONCLUSÕES: Os resultados obtidos via cálculos B3LYP/TZVP indicam que a reação de formação de alilamidas 5a e 5b provavelmente ocorre via um mecanismo do tipo SN1 por passos, com formação de um intermediário carbocátion o qual pode ser sustentado por dados experimentais cinéticos e os estudos de Relação Linear de Energia Livre de Hammett. Também se observou que há uma competição entre os mecanismos SN1 e SN1, porém dados experimentais mostram uma preferência ao mecanismo SN1, pois o produto majoritário da reação é a alilamida 5b.
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA: BORDWELL, F. G. Acc. Chem. Res. 1970, 3, 281.
CAREY, F. A.; SUNDBERG, R. J. Advanced Organic Chemistry; Springer Verlag: New York, 2007; 5th ed, pp 253-272.
COELHO, F.; ALMEIDA, P. W. Química Nova 2000, 23, 98.
FAGGION, D.J. Estudos Cinéticos das Reações de Formação de Acetamidas Alilicas e Bromação de Adutos de Morita-Baylis-Hillman Trabalho de Conclusão de Curso, Centro de Ciências Físicas e Matemáticas, Departamento de Química, UFSC. 55p Florianópolis- SC 2009.
FERREIRA, M.; FERNANDES, L.; SÁ, M. M. J. Braz. Chem. Soc. 2009, 20, 564.
MONK, J. P.; BROGDEN, R. N. Drugs 1991, 659.
POPLE, J. A.; et al, Gaussian 03, Revision B.03, Gaussian, Inc., Pittsburgh PA, 2003.
SINGH, V.; BATRA, S. Tetrahedron 2008, 64, 4511.
