ÁREA: Química Analítica
TÍTULO: DESENVOLVIMENTO E VALIDAÇÃO DE MÉTODO DE QUANTIFICAÇÃO DE IVERMECTINA EM MEDICAMENTOS VETERINÁRIOS POR CROMATOGRAFIA LÍQUIDA DE ALTA EFICIÊNCIA
AUTORES: ARANTES, J. F. M. (UNICAMP) ; MORAIS, L. S. R. (UNICAMP) ; RIBEIRO, C. C. (UNICAMP) ; RATH, S. (UNICAMP)
RESUMO: Neste trabalho foi desenvolvido e validado método para quantificação de ivermectina em medicamentos veterinários por cromatografia líquida de alta eficiência com detecção por arranjo de diodos (HPLC-DAD). Os parâmetros cromatográficos para o método foram otimizados conforme as especificações da monografia da Farmacopéia Britânica. A validação foi realizada conforme a Resolução n° 899, de 29 de maio de 2003 da ANVISA em amostras contendo ivermectina 1% em solução injetável. O método foi seletivo e os parâmetros de validação foram: linearidade: 0,9995, faixa linear de trabalho: 40-60 μg mL-1, precisão intra-dia: 0,21 - 1,1 %; precisão inter-dia: 1,3 2,7 % e exatidão: 96 100,9 %. Todos os parâmetros estão em conformidade com o preconizado pela ANVISA.
PALAVRAS CHAVES: ivermectina, validação, medicamentos veterinários
INTRODUÇÃO: O controle profilático e terapêutico de zoonoses foi apoiado em estratégias de manejo integrado à sanidade dos rebanhos, e envolveram, entre outras práticas, administração de fármacos sob forma injetável ou adicionados às rações. A ivermectina (IV) é um potente antiparasitário, derivado semi-sintético da avermectina B1, composto em média por 80 % de IV B1a e 20 % de IV B1b. Pela comprovada atividade endectocida é aplicada em gados, suínos, eqüinos, caprinos e ovinos e, dentre as avermectinas usadas como drogas, é uma das mais recorrentes (DANAHER et al., 2006). Pelos benefícios desses fármacos ao desempenho dos animais, sua recomendação e uso vêm crescendo no Brasil, mas questiona-se sua eficácia, já que o desempenho dos animais manejados sob condições ideais raramente é obtido. O Brasil demanda programas que garantam a qualidade de alimentos desta origem devido à expressiva atividade pecuária no cenário mundial. Cabe citar que o monitoramento de resíduos de fármacos em alimentos já é realizado pelo Ministério da Agricultura Pecuária e Abastecimento (MAPA), sendo fundamental para alavancar exportações de alimentos a países que definem tais práticas como requisito à negociação. Todavia, os medicamentos veterinários registrados no MAPA não são analisados em nenhum laboratório oficial e a avaliação da conformidade dos produtos nacionais ainda não é realizada. Considerando que alterações nas concentrações declaradas desses produtos comerciais possam implicar em perdas expressivas na indústria animal e comprometer a oportunidade de negociação internacional, este trabalho visa desenvolver e validar um método para quantificação de IV por HPLC-DAD em medicamentos veterinários para posterior aplicação em análises de medicamentos registrados no MAPA.
MATERIAL E MÉTODOS: Estabilidade: foi avaliada pela análise cromatográfica de soluções 100 μg mL-1 do padrão IV estocadas até 3 meses sob refrigeração (4 °C) e em temperatura ambiente (frascos transparente e âmbar).
Preparo de soluções: Soluções de IV foram preparadas no intervalo de 10 a 100 μg mL-1 por diluição da solução estoque do padrão de IV B1a 99 % Sigma-Aldrich a 1000 μg mL-1 em metanol (MeOH) grau HPLC. As amostras foram diluídas em MeOH, sonicadas por 30 minutos e filtradas em filtro de seringa 0,22 μm anterior análise cromatográfica.
Análise cromatográfica: As análises foram realizadas em um cromatógrafo à liquido 1525 (WATERS), coluna Purospher® STAR RP-18e (MERCK), 55 x 4,0 mm, 3 μm, e detecção a 245 nm. A eluição dos compostos foi isocrática sob temperatura de 30 ºC. A composição da fase móvel foi MeOH:H2O (83:17, v/v), vazão 1,0 mL min-1. Os parâmetros cromatográficos foram otimizados conforme especificações contidas na Farmacopéia Britânica (BRITISH PHARMACOPOEIA, 2009), resolução (Rs) superior a 3,0, fator de assimetria (As) de no máximo 2,5 e razão IV B1a/IV B1 maior que 90 % para o pico principal.
Validação: foram avaliados os seguintes parâmetros: linearidade, 10 a 100 μg mL-1; intervalo da faixa linear, 40 a 60 μg mL-1; precisão (intra-dia, n = 6 e inter-dia, n = 12), 50 μg mL-1; exatidão através do teste de recuperação em dois níveis de fortificação; e seletividade, avaliada através da presença de possíveis produtos de degradação ou compostos presentes na formulação. Os produtos de degradação da IV foram avaliados a partir da exposição do composto em meio ácido (HCl 0,1 mol L-1), básico (NaOH 0,1 mol L-1) , oxidante (H2O2 3 %) e sob temperatura (60 °C).
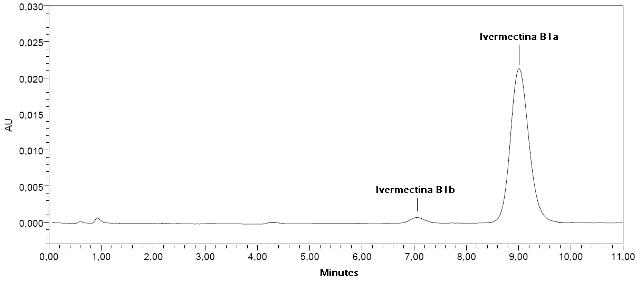
RESULTADOS E DISCUSSÃO: As soluções do padrão de IV apresentaram estabilidade por um período de no mínimo 3 meses quando estocadas a temperatura ambiente ou refrigeração. O composto não degrada na presença de luz nestas condições de armazenamento. Os parâmetros de conformidade do sistema cromatográfico obtidos pela eluição de IV sob as condições otimizadas estão apresentados na Tabela 1 e um cromatograma característico está apresentado na Figura 1.
Figura 1. Cromatograma da separação da IV B1a de seu homólogo B1b. Vazão 1,0 mL min-1, temperatura 30 ºC e λ = 245 nm. Fase móvel MeOH:H2O (83:17, v/v) e fase estacionária Purospher® STAR RP-18e.
Tabela 1. Parâmetros de conformidade do sistema cromatográfico.
A IV é estável em meio oxidante (H2O2 3%) e temperatura de 60 °C, durante 24 horas e instável em HCl e NaOH 0,1 mol L-1. No entanto, nenhum produto de degradação elui nos tempos de retenção da IV B1a e B1b, confirmando a seletividade do método cromatográfico desenvolvido. Também não foi verificada co-eluição de outros compostos das formulações injetáveis.
O método foi validado para 8 amostras de medicamentos de fabricantes diferentes, sendo que três apresentaram efeito matriz e, em conseqüência, a recuperação foi abaixo da faixa aceitável de 95 a 105%. O teor de IV nos medicamentos que apresentam efeito matriz precisam ser determinados pelo método de adição de padrão.
Os parâmetros de validação para os medicamentos que não apresentaram efeito matriz foram: linearidade: 0,9995, faixa linear de trabalho: 40-60 μg mL-1, precisão intra-dia: 0,21 - 1,1 %; precisão inter-dia: 1,3 2,7 % e exatidão: 96 100,9 %. Todos os parâmetros estão em conformidade com o preconizado pela ANVISA (ANVISA, 2003).

CONCLUSÕES: Os resultados obtidos estão em conformidade com as exigências das aplicações analíticas, segundo as recomendações da ANVISA, e de acordo com as orientações da monografia oficial 1336 da farmacopéia britânica. O método analítico proposto empregando a HPLC-DAD atende aos seus propósitos, assegurando a confiabilidade dos resultados. Ainda, o método apresenta vantagens quanto à simplicidade no preparo de amostra, constituição da fase móvel e baixo tempo de análise.
AGRADECIMENTOS: À Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) e ao Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
REFERÊNCIAS BIBLIOGRÁFICA: Agência Nacional de Vigilância Sanitária (ANVISA). Guia para Validação de Métodos Analíticos e Bioanalíticos, RE nº 899, de 29/05/2003.
British Pharmacopoeia. Monographs: Medicinal and Pharmaceutical Substances: Ph Eur Monograph 1336. V. I & II, p. 3298-3304, 2009.
Danaher, M., Holwells, L. C., Crooks, S. R. H., Cerkvenik-Flajs, V., O'Keeffe M. Journal of Chromatography B. V.844, p.175-203, 2006.
