ÁREA: Físico-Química
TÍTULO: ESTUDO TEÓRICO QUÍMICO - QUÂNTICO DO COMPLEXO DIBENZOTETRAAZA[14]ALUNENO DE FERRO (II).
AUTORES: COSTA, H. R. (UFMA) ; BRITO, J. G. L. (UFMA) ; SILVA, A. L. P. (UFPB) ; VARELA JÚNIOR, J. J. G. (UFMA) ; TANAKA, A. A. (UFMA)
RESUMO: Este trabalho apresenta estudos computacionais do complexo [Fe(II)TAA] usando cálculos
ab initio com base na teoria funcional da densidade (DFT). Sendo assim, a molécula de
Fe(II)TAA, com várias multiplicidades, foi otimizada e um estudo comparativo com dados
experimentais foi realizado, a fim de investigar as distâncias e ângulos de ligação,
bem como outros parâmetros, como, a energia total e os orbitais de fronteira (LUMO-
HOMO). A menor energia total, encontrada no complexo Fe(II)TAA, mostrou que o estado
tripleto é o mais estável, embora o estado esperado, para anéis complexos do tipo
quadrado planar com centro metálico de metais de transição da primeira fila, fosse
singleto (spin baixo). Tal anomalia é resultado do efeito de distorção tetragonal (ou
efeito Jahn-Teller).
PALAVRAS CHAVES: dibenzotetraaza[14]aluneno; teoria funciona da densidade; efeito jahn-teller.
INTRODUÇÃO: O estudo das estruturas cristalinas e moleculares dos complexos de metais de
transição, especialmente os sintéticos com ligantes macrocíclicos, torna-se
necessária à compreensão de suas propriedades físicas e químicas. Neste contexto, os
tetraazamacrociclos ao se coordenarem a íons metálicos conferem a estes centros de
coordenação uma estabilização, devido ao chamado "efeito macrocíclico", relacionado à
conjugação do sistema no que diz respeito à expansão e contração radial dos doadores
do N4, juntamente com as exigências estéricas do metal íons com raios variados, como
Ferro, Cobalto, Manganês e Níquel, pode levar a coordenação com alta reatividade [1-
3]. Além disso, a planaridade destes complexos altamente conjugados relaciona-se com
as interações intermoleculares semelhantes aos complexos quadrado-planares de quatro
coordenadas, como complexos de dimetilglioxima de Ni(II) [4,5].
Assim, o objetivo do presente trabalho é realizar um estudo teórico químico-quântico
do complexo Fe(II)TAA em nível de DFT, com o auxílio do programa Gaussian 09 [6], com
o intuito de verificar a estrutura mais estável e compará-la com obtido
experimentalmente [7].
MATERIAL E MÉTODOS: O estudo teórico foi conduzido por meio de cálculos ab initio de estrutura
eletrônica. A otimização das geometrias das estruturas, além dos cálculos de energia
de ligação e distribuição de cargas da Fe(II)TAA, foi realizada com a utilização do
programa GAUSSIAN 09 [6]. Foram empregados cálculos baseados na teoria do funcional
da densidade (DFT), utilizando o funcional híbrido de três parâmetros de Becke com as
correções de gradiente fornecidas pelos funcionais de Lee, Yang e Parr (B3LYP) [8].
As funções de bases LANL2DZ (átomo de Ferro) e 6-31G* (átomos de carbono, nitrogênio
e Hidrogênio) foram utilizadas para otimizacão das estruturas, análise dos orbitais e
descrição das frequências vibracionais.
A avaliação da estrutura eletrônica foi feita através do método NBO (Natural Bond
Orbitals) [9], que localiza os orbitais canônicos e os transforma em orbitais de
centro, orbitais de ligação, orbitais de caroço e de pares isolados, de acordo com a
visão de estrutura química de Lewis. Os NBOs são compostos de orbitais híbridos, os
quais são combinações lineares de orbitais atômicos naturais em um dado centro. Além
disso, os valores da densidade, ρ(r), do Laplaciano, ∇2ρ(r), e da
eliplicidade, ε,
serão empregados para melhor avaliar as variações das densidades eletrônicas e a
natureza da ligação química.
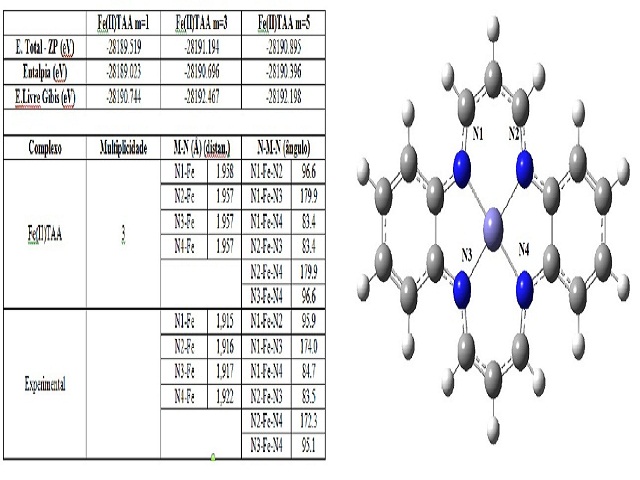
RESULTADOS E DISCUSSÃO: Na Figura 1, onde mostramos os resultados obtidos com várias multiplicidades de spin,
observamos que o estado mais estável foi tripleto, pois complexos quadrado-planares
apresentam uma grande distorção tetragonal em orbitais eg com quebra de
degenerescência entre os orbitais dx2 y2 e dz2. Ou seja, esperava-se que nesse
complexo o estado de spin baixo (singleto) fosse predominante. No entanto, o que
aconteceu foi uma distorção nos orbitais dxy com quebra de degenerescência entre os
orbitais dxz e dyz (efeito Jahn-Teller) [10]. Então, a diferença energética entre o
tripleto e o quinteto é de 6,9 kcal/mol. Já a diferença de energia entre o estado
esperado singleto e o observado tripleto é de 38,6 kcal/mol. Dessa forma, tal
diferença pode ser explicada pela análise dos orbitais naturais, obtida pelo método
NBO.
As distâncias e os ângulos de ligação para o tripleto foram medidos e comparados com
resultados experimentais [7]. As distâncias Fe-N calculadas, superestimam a
experimental em aproximadamente 0,04 Å, já a variação dos ângulos é baixa, sendo
superior ao experimental em apenas 0,7 º, mostrando boa concordância com os dados.
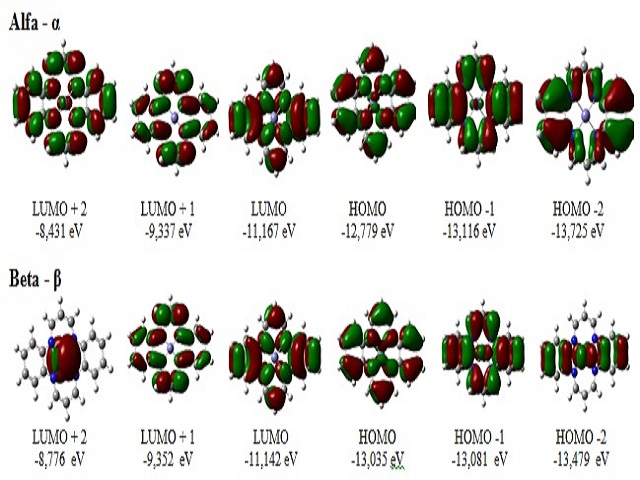
A Figura 2 mostra a representação esquemática dos orbitais LUMO, LUMO+1 e LUMO+2 e o
dos HOMO, HOMO-1 e HOMO-2, para os elétrons α e β do Fe(II)TAA. Observou-se
uma
considerável densidade de carga formada no centro metálico do HOMO para os elétrons
α
e β, porém nenhuma densidade de carga é observada no LUMO dos elétrons α e
β. Sendo
assim, esta molécula apresenta um caráter nucleofílico que é representado pela
densidade de carga do HOMO e seu alto valor enérgico [9].
CONCLUSÕES: Os resultados dos cálculos através de DFT, mostraram ser satisfatórios para estudar as
propriedades geométricas e energéticas do complexo de Fe(II)TAA, visto que, se obteve
uma boa aproximação das distâncias e ângulos de ligação em comparação com dados
experimentais. Neste sentindo, também foi possível observar, através dos orbitais de
fronteira (LUMO-HOMO), o caráter nucleofílico deste complexo, indica assim uma possível
característica catalítica para estudos de reação de moléculas como, por exemplo, a
reação de redução de oxigênio.
AGRADECIMENTOS: Os autores agradecem a FAPEMA e a UFMA pelo suporte técnico concedido.
REFERÊNCIAS BIBLIOGRÁFICA: [1] SISTER, E. et al. Inorganic Chemistry, v. 27, n. 4, p. 600-604, 1988.
[2] WEISS, M. C.; GORDON, G.; GOEDKEN, V. L. Inorganic Chemistry, v. 16, n. 2, p. 305-310, 1977.
[3] WEISS, M. C. et al. Journal of the American Chemical Society, v. 98, p. 8021-8031, 1976.
[4] AZUMA, N. et al. Journal of the Chemical Society, v. 1, p. 343-348, 1995.
[5] GIROLAMI, G. S.; RAUCHFUSS, T. B. ANGELICI, R. J. Synthesis and Technique in Inorganic Chemistry: A Laboratory Manual. 3. ed., 1999.
[6]. FRISCH, M. J. et al. Gaussian 03, Revision B.04; Gaussian, Inc.: Pittsburgh PA, 2003.
[7] VIRGIL, L. et al. Journal of the American Chemical Society, v. 98, p. 8014-8021, 1976.
[8] LEE, C.; YANG, W.; PARR, R. G. Physical Review B, v. 37, p. 785-789, 1988.
[9] CARPENTER, J.E.; WEINHOLD, F.J. Journal of Molecular Structure, v. 169, p. 41-48, 1988.
[10] LEE, J. D. Química inorgânica: não tão concisa 5. ed., 1999.
