ÁREA: Físico-Química
TÍTULO: ESTUDO EM NÍVEL DE DFT DO COMPLEXO [Ni(TAA)]2+ DIBENZOTETRAAZA[14]ANULENO DE NÍQUEL (II).
AUTORES: SILVA, A. L. P. (UFPB) ; COSTA, H. R. (UFMA) ; VARELA JÚNIOR, J. J. G. (UFMA) ; TANAKA, A. A. (UFMA) ; MARQUES, A. L. B. (UFMA)
RESUMO: Neste trabalho desenvolveu-se um estudo teórico-quântico da estrutura geométrica e da
energia do estado mais estável, bem como dos orbitais moleculares para o complexo
[Ni(TAA)]2+ em nível da teoria do funcional da densidade (DFT). A geometria do complexo
foi otimizada em simetria D2h, com o níquel coordenado a quatro átomos de nitrogênio.
Os parâmetros geométricos (distância e ângulo de ligação) da estrutura teórica estão de
acordo com obtidos experimentalmente, além disso, a multiplicidade de spin, singleto,
foi calculada como a mais estável para o complexo. As cargas de Mulliken mostraram-se
bem distribuídas sobre os nitrogênios, em torno de -0,53, e os orbitais de fronteira
(HOMO-LUMO) bem deslocalizados, sendo que o níquel só participa da formação do HOMO,
com um gap de 1,60 eV.
PALAVRAS CHAVES: química quântica, teoria do funcional da densidade, complexo de níquel.
INTRODUÇÃO: O contínuo interesse na química dos complexos tetraazamacrocíclicos é em grande parte
devido à sua semelhança estrutural com sítios ativos das porfirinas e
metaloporfirinas, e sua relevância em sistemas supramoleculares e ciência dos
materiais [1]. Entre os macrociclos sintéticos, os sistemas que contêm a
dibenzotetraaza[14]anulene, tem sido estudado extensivamente nos últimos 40 anos,
desde a primeira preparação em 1969 por Jager e Anorg [2], e foram apreciados tanto
por sua acessibilidade sintética e química muito rica. Conquistas mais importantes
nesta área têm sido publicado recentemente [3], incluindo a síntese e estrutura de
derivados de dibenzotetraaza[14]anulene com metais de transição. Assim, o
dibenzotetraaza[14]anulene parece ser especialmente atraente, já que é conhecido por
ser altamente reativa nas posições meso do anel macrocíclico [4] e, portanto, bem
adequado para o modificação da estrutura. Além disso, eles adotam a forma não-planar,
fornecendo assim a base de uma cavidade molecular para uma inclusão do metal.
Portanto, parece razoável esperar, que a introdução de metais selecionados na
arquitetura molecular do ligante permitiria induzir uma forma de seletividade para a
complexação de metais, e, consequentemente, qualquer reação axial dos complexos de
metal correspondente [5].
Logo, diante do exposto, o objetivo deste trabalho é realizar um estudo químico-
quântico do complexo [Ni(TAA)]2+ em nível de teoria do funcional da densidade, com o
auxílio do programa Gaussian 03 [6], com o intuito de verificar a estrutura mais
estável e compará-la com obtido experimentalmente [7].
MATERIAL E MÉTODOS: Neste trabalho, a análise conformacional das estruturas iniciais do complexo
[Ni(TAA)]2+, bem como os cálculos de otimização estrutural da geometria baseados na
teoria do funcional da densidade. A otimização dos parâmetros geométricos (distância
e ângulo de ligação) e da simetria das estruturas do complexo [Ni(TAA)]2+, além dos
cálculos de energia total, distribuição de cargas e orbitais de fronteira o HOMO
(Orbital Molecular Ocupado de mais alta Energia) e o LUMO (Orbital Molecular
desocupado de mais baixa energia), foram realizadas com a utilização do programa
GAUSSIAN 03, Revision B04 [6]. Foram empregados cálculos baseados na teoria do
funcional da densidade (DFT), utilizando o funcional híbrido de três parâmetros de
Becke com as correções de gradiente fornecidas pelos funcionais de Lee, Yang e Parr
(B3LYP) [8]. As funções de bases LANL2DZ (usada para o átomo de níquel), 6-31G*
(usada para os átomos de carbono, nitrogênio e hidrogênio). Os cálculos de
frequências vibracionais das estruturas otimizadas foram usados para gerar as
correções de energia vibracional do ponto zero (ZPVE).
A avaliação das possíveis multiplicidades de spin (2S+1) para o complexo foram
testadas, com intuito de verificar diferenças energéticas, entre as multiplicidades
singleto e tripleto para o complexo [Ni(TAA)]2+, ademais as estruturas eletrônicas
foram realizadas através do método NBO (Natural Bond Orbitals). A análise dos
Orbitais de NBO foi conduzida de maneira a encontrar as propriedades eletrônicas da
estrutura mais estável para o complexo [Ni(TAA)]2+.
RESULTADOS E DISCUSSÃO: A partir dos dados de simetria obtida pelo Gaussian 03 [6] constatou-se que a
molécula apresenta simetria D2h, com o níquel ligado aos quatro nitrogênios do
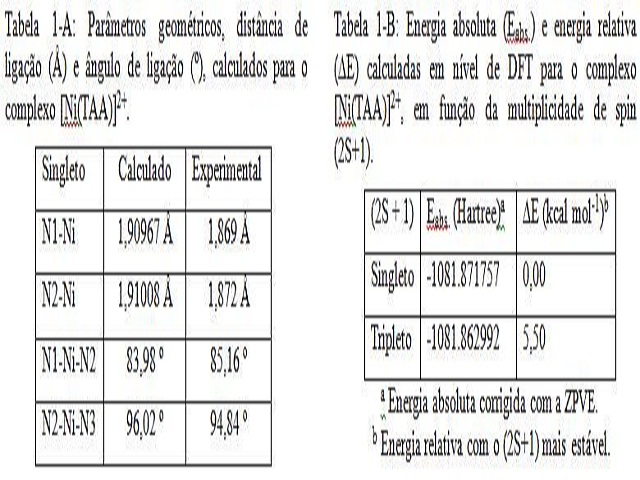
macrociclo, além disso, analisando a Tabela 1-A, vide Figura 1, observa-se que os
parâmetros geométricos para o complexo [Ni(TAA)]2+, estão em bom acordo com os
resultados experimentais [7], sendo que a distância teórica superestima a
experimental em 0,04 Å, desviando em 2,14 %, e os ângulos variam entre ±1,18 º,
desviam em 1,38 %, indicando uma leve distorção no complexo. Já com relação à
multiplicidade de spin, para o Ni2+, com configuração d8, testou-se o singleto e o
tripleto, vide Tabela 1-B, vide Figura 1, sendo que o singleto é mais estável em 5,50
kcal mol-1, isto se deve ao fato do nitrogênio ser um ligante campo forte [9].
A análise populacional do orbital molecular ou cargas de Mulliken mostrou que a carga
do Ni era de +0,62, enquanto as cargas sobre os quatro nitrogênios estavam bem
distribuídas, em torno de -0,53, indicando um forte caráter iônico na ligação Ni-N.
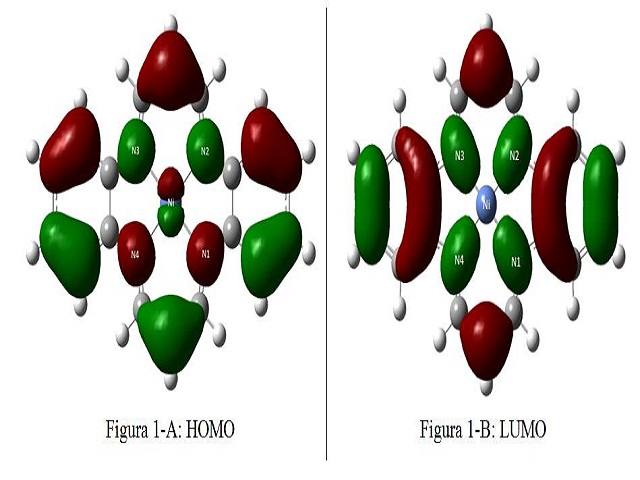
Além disso, a análise dos orbitais moleculares de fronteira o HOMO e o LUMO, que
constitui uma ferramenta favorável para o entendimento das ligações químicas entre o
centro metálico e os ligantes, conforme mostrado nas Figuras 2-A e 2-B, vide Figura
2, observa-se que os orbitais encontram-se bem deslocalizados sobre a molécula, e que
não há participação efetiva do níquel na formação do LUMO, ademais o baixo gap (HOMO-
LUMO) de 1,60 eV, confirmando a natureza da ligação iônica entre o níquel e os quatro
nitrogênios do ligante.
CONCLUSÕES: Neste trabalho constatou-se em nível DFT que o complexo [Ni(TAA)]2+, apresentou
simetria otimizada D2h, com o Ni coordenado a quatro átomos de N. Os parâmetros
geométricos da estrutura proposta, estão de acordo com os parâmetros obtidos
experimentalmente, e a multiplicidade de spin, singleto, foi calculada como sendo a
mais estável. Além disso, as cargas de Mulliken mostraram-se bem distribuídas sobre os
quatro átomos N, em torno de -0,53, os orbitais de fronteira (HOMO-LUMO) mostaram-se
bem deslocalizados sobre o complexo, com um gap de 1,60 eV.
AGRADECIMENTOS: Os autores agradecem a SEDUC-MA, a FAPEMA e a UFMA, pelo suporte técnico concedido.
REFERÊNCIAS BIBLIOGRÁFICA: [1] MOUNTFORD, P. Dibenzotetraaza[14]annulenes: versatile ligands for transition and main group metal chemistry. Chemical Society Reviews, v. 27, p. 105-115, 1998.
[2] EILMES, J.; MICHALSKI, O.; WOZNIAK, K. New chiral receptors based on dibenzotetraaza[14]annulenes. Inorganica Chimica Acta, v. 317, p. 103-113, 2001.
[3] GLOLIK, J. et al. Lacunar derivates of dibenzotetraaza[14]annulene: synthesis and crystal structure of new receptors produced via bis-alkylation of the macrocyclic precursor. Tetrahedron, v. 67, p. 2623-2632, 2011.
[4] AZUMA, N. et al. A crystal modification of dibenzo[1,4,8,11]tetraaza[14]annulene: X-ray molecular structure and proton tautomerism of the highly л-conjugated form. Journal of the Chemical Society, v. 1, p. 343-348, 1995.
[5] GAWINKOWSKI, S.; EILMES, J.; WALUK, J. Structure, vibrations, and hydrogen bond parameters of dibenzo[1,4,8,11]tetraaza[14]annulene nickel(II). Journal of Molecular Structure, v. 976, p. 215-225, 2010.
[6] FRISCH, M. J. et al. Gaussian 03, Revision B.04; Gaussian, Inc.: Pittsburgh PA, 2003.
[7] WEISS, M. C.; GORDON, G.; GOEDKEN, V. L. Crystal and molecular structure of the macrocyclic nickel(II) complex Ni(C18H14N4): dibenzo[1,4,8,11]tetraaza[14]annulene nickel(II). Inorganic Chemistry, v. 16, n. 2, p. 305-310, 1977.
[8] BECKE, A. D., Density-functional Exchange-energy approximation with correct asymptotic behavior. Physical Review A, v. 38, p. 3098-3100, 1988.
[9] HUHEEY, J. E.; KEITER, E. A.; KEITER, R. L. Inorganic chemistry: principles structure and reactivity. 4. ed. New York: Haper Collins, 1993.
