ÁREA: Físico-Química
TÍTULO: ANÁLISE COMPUTACIONAL DOS PRODUTOS ISOMÉRICOS DE CADEIA DA REAÇÃO DE HALIDRIFICAÇÃO EM ALCENOS
AUTORES: LOBATO, C.C. (UEAP) ; TRINDADE, R.D. (UEAP) ; DINIZ, J.E.M. (UEAP) ; SANTOS, C.B.R. (UNIFAP)
RESUMO: A química computacional na atualidade é uma ferramenta cada vez mais útil e desejável para o ensino de química e pesquisa, sendo possível obter resultados altamente confiáveis de cálculos de propriedades químico-quânticas no estudo de uma ampla faixa de problemas de interesse químico. Este trabalho teve como objetivos analisar a estabilidade de produtos isoméricos de cadeia a partir de reações de halidrificação em alcenos, com auxílio de cálculos computacionais; interpretação dos mapas de potencial eletrostático dos substratos, reagentes e produtos; motivar os acadêmicos de química e áreas afins a investigar os conceitos de química quântica, físico-química e química orgânica através de modelagem molecular.
PALAVRAS CHAVES: reação de halidrificação; química quântica e métodos computacionais.
INTRODUÇÃO: Devido aos recentes progressos da área computacional e no desenvolvimento de eficientes algoritmos de cálculos, um grande avanço foi verificado no desenvolvimento dos cálculos químico-quânticos moleculares. Métodos ab initio, DFT e semi-empíricos fornecem parâmetros realísticos em curto tempo. A Química Quântica fornece uma descrição mais acurada e detalhada dos efeitos eletrônicos quando comparada aos métodos empíricos (ARROIO et al, 2010).
O tratamento teórico mais rigoroso não faz uso de parâmetros empíricos e é denominado ab initio. Embora este tipo de método forneça informação relativamente mais precisa sobre o comporta¬mento eletrônico, ele é, em termos operacionais, lento e mais caro. Por isso, vários métodos semi-empíricos foram desenvolvidos, os quais são baseados em algumas suposições que servem para sim¬plificar os cálculos e utilizam certos parâmetros obtidos dos dados experimentais. Vale ressaltar que a precisão desses métodos está relacionada ao erro associado ao conjunto de base selecionado e ao nível de tratamento da correlação eletrônica.
A química computacional na atualidade é uma ferramenta cada vez mais útil e desejável para o ensino de química e pesquisa, sendo possível obter resultados altamente confiáveis de cálculos de propriedades químico-quânticas no estudo de uma ampla faixa de problemas de interesse químico (Hessley, 2000). Este trabalho tem como objetivos analisar a estabilidade de moléculas a partir de reações de halidrificação em alcenos com auxílio de cálculos computacionais; interpretar os mapas de potencial eletrostático dos reagentes, substratos e produtos; motivar os acadêmicos de química e áreas afins a investigar os conceitos de química quântica, físico-química e química orgânica através de modelagem molecular.
MATERIAL E MÉTODOS: A proposta foi desenvolvida para reações de halidrificação utilizando dois substratos (2-buteno e 2-metilpropeno) e quatro reagentes nucleofílicos (HF, HCl, HBr e HI), para analisar a estabilidade conformacional dos isômeros de cadeia dos halogênios (C4H9F, C4H9Cl, C4H9Br e C4H9I). Os substratos, reagentes e produtos foram construídos com auxílio do programa HyperChem Release 6.02 e otimizados com diferentes níveis de teoria (semi-empíricos AM1/PM3, ab initio HF/3-21G e DFT 3-21G), utilizando o sistema GNU/Linux. Os parâmetros energéticos, energia total, energia nuclear e energia eletrônica foram utilizadas para correlacionar estrutura e a estabilidade da molécula. A energia total foi utilizada para estimar a estabilidade dos produtos, e corresponde a soma da energia de repulsão nuclear com a energia eletrônica. A energia eletrônica foi determinada mediante a aproximação de Born-Oppenheimer assumindo-se uma posição fixa dos núcleos, e a equação de Schrödinger foi resolvida a fim de se encontrar a energia eletrônica das moléculas. A energia do orbital molecular ocupado de mais alta energia (εHOMO), energia do orbital molecular desocupado de energia mais baixa (εLUMO), diferença de energia entre os orbitais HOMO e LUMO (∆ε), comprimento de ligação na posição C2-X (Å), carga atômica líquida nos átomos C2 e X nos produtos isoméricos (qC2 e qx), momento de dipolo molecular (µ), dureza (η), moleza (S), propriedades QSAR como: Volume (V), Energia de Hidratação (HE), Coeficiente de partição octano-água (Log P), Refratividade (RM), polarizabilidade molecular (α), área da superfície (SA), massa molecular (m) e os mapas de potencial eletrostático molecular (MEP), foram calculados para os substratos, reagentes e produtos da reação de halidrificação.
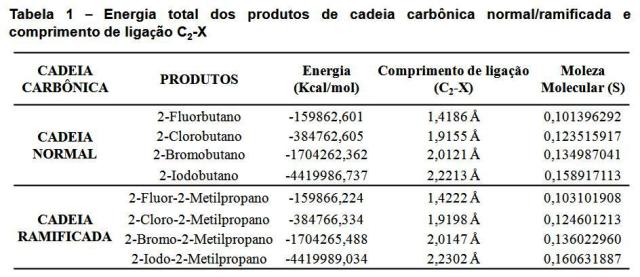
RESULTADOS E DISCUSSÃO: Para validar o nível de teoria foi calculada a energia total para os substratos (2-buteno e 2-metil-propeno), com a finalidade de obter a menor energia do sistema e a maior estabilidade conformacional. O método ab initio 3-21G/HF apresentou menor energia para ambos os substratos, sendo que para o 2-Buteno a energia foi de -97414,94241 kcal/mol. O método ab initio 3-21G/HF quando comparado com os demais métodos computacionais, variou da ordem de 14,357% em relação ao DFT, 85,269% em relação ao AM1 e 85,859% em relação ao PM3; e para 2-metil-propeno apresentou energia de -97414,39089 kcal/mol variando da ordem de 14,356% em relação ao DFT, 85,272% em relação ao AM1, 85,859% em relação ao PM3. Após a análise das energias dos substratos os produtos isoméricos das reações foram calculados e otimizados utilizando o método ab initio 3-21G/HF.
Dentre os produtos das reações entre 2-metil-propeno e os haletos de hidrogênio, o composto que apresentou menor energia e maior estabilidade foi o 2-Iodo-2-Metilpropano com energia de -4419989,034 kcal/mol, em relação ao mais estável o 2-Bromo-2-Metilpropano, 2-Cloro-2-Metilpropano e 2-Fluor-2-Metilpropano obteve-se energias da ordem de 38,558%, 8,705% e 3,616%, respectivamente, ver Tabela 1.
A Figura 1 mostra o MEP para o composto mais (I) e menos (II) estável dentre os investigados. Notamos o potencial eletrostático negativo (região vermelha), na dupla ligação do substrato e nos halogênios dos reagentes. No produto mais estável temos uma distribuição homogênea na parte hidrofóbica, devido apresentar (V), (HE) e LogP igual a 392,79Å3, 2,30kcal/mol e 2,19, respectivamente. Para o produto menos estável há uma distribuição de potencial positiva na parte hidrofóbica e uma região negativa no átomo de flúor caracterizando a parte hidrofílica.
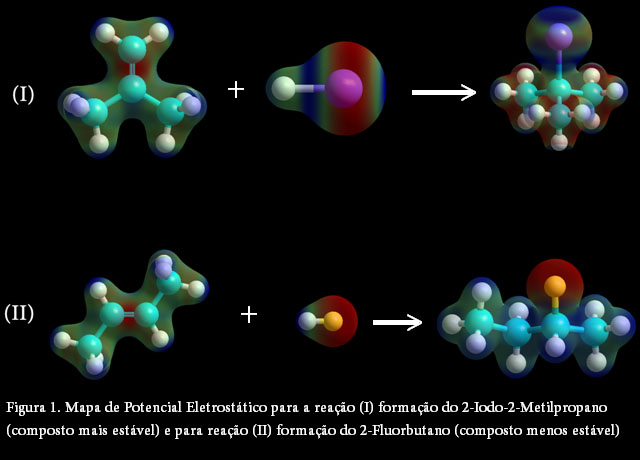
CONCLUSÕES: O uso de modelagem computacional deve ser incorporado nas grades curriculares, pois possibilita abordar um número maior de tópicos e um aprofundamento da teoria. Após validar o melhor nível de teoria 3-21G/HF para os substratos afim de obter a menor energia do sistema e maior estabilidade conformacional. O composto mais estável foi o 2-Iodo-2-Metilpropano, quando comparados com as energias dos compostos isoméricos analisados. O MEP mostrou que o composto mais estável tem maior tendência de formar carbocátion terciário, devido o átomo de iodo ter maior raio atômico e menor energia de ionização.
AGRADECIMENTOS: Universidade Federal do Amapá - UNIFAP
Ao Laboratório de Fármacos pela Infra-Estrutura Computacional
Universidade do Estado do Amapá -
REFERÊNCIAS BIBLIOGRÁFICA: Arroio, A.; Honório, K. M.; Silva, A. B. F. Propriedades químico-quânticas empregadas em estudos das relações estrutura-atividade. Química Nova, Vol. 33, No. 3, 694-699. 2010
CHEMPLUS: Modular Extensions for HyperChem Release 6.02, Molecular Modeling for Windows, HyperClub, Inc., Gainesville, 2000.
Hessley, R. K. J. Chem. Educ. 2000, 77, 794.
