ÁREA: Físico-Química
TÍTULO: ANÁLISE DOS ORBITAIS NATURAIS DE LIGAÇÃO DO COMPLEXO TRICLORO[2,4,6-TRIS(2-PIRIDIL)-1,3,5-TRIAZINA] RÓDIO (III), [Rh(TPTZ)Cl3].
AUTORES: SILVA, A. L. P. (UFPB) ; VARELA JÚNIOR, J. J. G. (UFMA) ; TANAKA, A. A. (UFMA) ; MARQUES, A. L. B. (UFMA)
RESUMO: Neste trabalho desenvolveu-se um estudo teórico da estrutura geométrica e dos orbitais
naturais de ligação, para o complexo [Rh(TPTZ)Cl3] em nível da teoria do funcional da
densidade (DFT), B3LYP/LANL2DZ. A geometria octaédrica otimizada do complexo foi em
simetria Cs, com o Ródio coordenado a três átomos de Nitrogênio e três de Cloro,
mostrou-se levemente distorcida. Os parâmetros geométricos da estrutura teórica estão
de acordo com obtidos experimentalmente, além disso, constatou-se, através da análise
de NBO, uma forte polarização nas ligações Rh-N e Rh-Cl, com o Rh atuando na ligação
entorno de 22%, e os orbitais de fronteira (HOMO-LUMO) mostraram-se bem deslocalizados,
apresentando um gap de 3,11 eV.
PALAVRAS CHAVES: orbitais naturais de ligação (nbo), teoria do funcional da densidade, tricloro[2,4,6-tris(2-piridil)-1,3,5-triazina] ródio (iii).
INTRODUÇÃO: Há um interesse de continuar os estudos do ligante 2,4,6-tris(2-piridil)-1,3,5-
triazina, o TPTZ, para ser usado como um reagente analítico para complexação de
diversos íons metálicos [1]. Nos últimos anos, o TPTZ que funciona simultaneamente
como um ligante bidentado e tridentado, ganhou considerável interesse devido ao seu
uso como um espaçador para projetar complexos supramoleculares. Estes complexos podem
ser usados em uma variedade de reações eletrocatalítica [2]. Os compostos da família
2,4,6-triazinas são geralmente estáveis à hidrólise, o ácido mineral concentrado e
temperaturas acima de 150 Cº são necessários para a sua reação de hidrólise. No
entanto, Julve et al [3] encontraram o Cu (II) em meio aquoso promovendo a hidrólise
do TPTZ para o bis (2 piridilcarbonil) amida; além disso também foram relatadas
caracterizações cristalográficas dos complexos de cobre com TPTZ hidrolisado. Já Paul
et al [4] estudaram os efeitos de vários solventes na síntese de diferentes complexos
de ródio, partindo de RhCl3 e TPTZ, caracterizando por cristalografia o complexo
[Rh(TPTZ)Cl3]. Holbrey et al [5] sintetizaram um complexo similar, com centro
metálico de cobalto (III), sintetizado e caracterizado por Raio-X.
Assim, o objetivo do presente trabalho é realizar um estudo químico-quântico em nível
de teoria do funcional da densidade, B3LYP/LANL2DZ, com o auxílio do programa
Gaussian 03 [6], com o intuito de analisar os orbitais naturais de ligação [7,8]
envolvidos no complexo [Rh(TPTZ)Cl3].
MATERIAL E MÉTODOS: Neste trabalho, a análise conformacional das estruturas iniciais do complexo
[Rh(TPTZ)Cl3], bem como os cálculos de otimização estrutural da geometria baseados na
teoria do funcional da densidade. A otimização dos parâmetros geométricos (distância
e ângulo de ligação) e da simetria das estruturas do complexo [Rh(TPTZ)Cl3], além dos
cálculos de energia total e orbitais de fronteira o HOMO (Orbital Molecular Ocupado
de mais alta Energia) e o LUMO (Orbital Molecular desocupado de mais baixa energia),
foram realizadas com a utilização do programa GAUSSIAN 03, Revision B04 [6]. Foram
empregados cálculos baseados na teoria do funcional da densidade (DFT), utilizando o
funcional híbrido de três parâmetros de Becke com as correções de gradiente
fornecidas pelos funcionais de Lee, Yang e Parr (B3LYP) [9]. A função de base LANL2DZ
(usada para os átomos de ródio, nitrogênio, cloro, carbono e hidrogênio). Os cálculos
de frequências vibracionais das estruturas otimizadas foram usados para gerar as
correções de energia vibracional do ponto zero (ZPVE).
A avaliação das estruturas eletrônicas foi realizada através do método NBO (Natural
Bond Orbitals). A análise dos Orbitais de NBO foi conduzida de maneira a determinar
as propriedades das ligações para o complexo [Rh(TPTZ)Cl3].
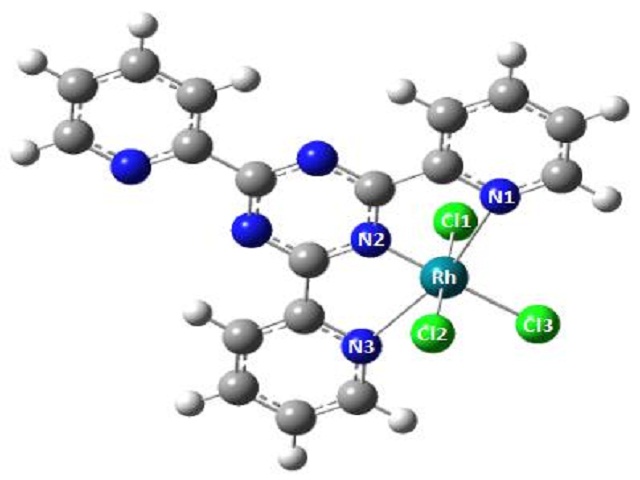
RESULTADOS E DISCUSSÃO: A partir da Figura 1, constatou-se que o complexo [Rh(TPTZ)Cl3] apresenta estrutura
otimizada com simetria Cs, com o Ródio coordenado a três átomos de Nitrogênio e três
de Cloro, que apresenta uma energia total de -2511,38 Hartree. Além disso, nas
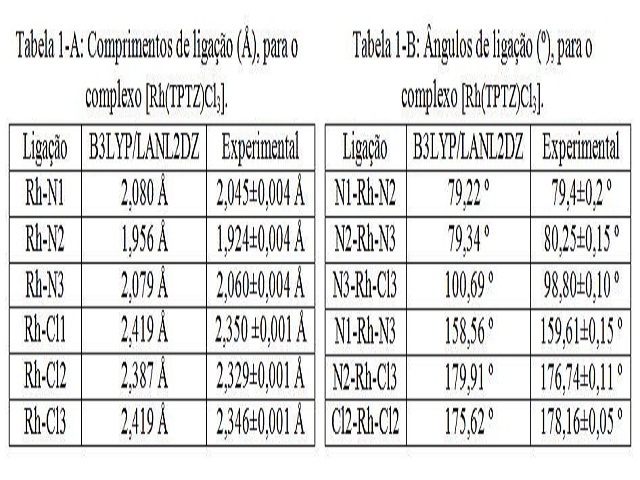
Tabelas 1-A e 1-B, vide Figura 2, são mostrados os parâmetros geométricos calculados
do complexo [Rh(TPTZ)Cl3] em nível B3LYP/LANL2DZ, e estes estão em bom acordo com os
resultados experimentais [4], sendo que a distância teórica superestima a
experimental em no máximo 1,66% para Rh-N e 3,86% para Rh-Cl, já os ângulos desviam
em 1,79 %, indicando assim, uma leve distorção no complexo, como observado
experimentalmente [4]. Já a análise dos orbitais de fronteira HOMO-LUMO, constatou-se
que estes são bem deslocalizados, com participação do centro metálico de Rh na
formação dos orbitais, além de apresentar um gap (HOMO-LUMO) de 3,11 eV.
A partir da análise de NBO, constatou-se ligação Rh-N está formada pela interação
entre o orbital híbrido [sp3,11d2,25] centrado no íon Rh(III), com o orbital híbrido
[sp3,12] localizado no átomo de nitrogênio. A ligação Rh-Cl está formada pela
interação entre um orbital [sp2,87d1,87] centrado sobre o ródio, e um orbital
[sp6,32] centrado no cloro. O grau de participação na formação da ligação Rh-N é de
21,16% para o Rh(III) e de 78,84% para o N. Já, para a ligação Rh-Cl, a participação
do Rh(III) é do 25,24%, e de 74,76% para o Cl. Esses resultados mostram uma
polarização elevada na direção dos átomos de N e Cl sobre as ligações Rh-N e Rh-Cl,
respectivamente, isto se deve a elevada eletronegatividade do N e do Cl [10].
CONCLUSÕES: Neste trabalho constatou-se em nível de DFT que o complexo octaédrico [Rh(TPTZ)Cl3],
apresentou simetria otimizada Cs, levemente distorcida. Os parâmetros geométricos da
estrutura teórica proposta, corroboram com os parâmetros obtidos experimentalmente.
Além disso, mostrou-se que a participação do Rh(III), na formação das ligações Rh-N e
Rh-Cl, foi cerca de 25%. Esses resultados mostram uma polarização elevada na direção
dos átomos de N e Cl. Finalizando, A análise de NBO indicou que a hibridização do
Rh(III) foi [sp3,11d2,25] concordante com a coordenação octaédrica prevista, [sp3d2].
AGRADECIMENTOS: Os autores agradecem a SEDUC-MA, a FAPEMA e a UFMA, pelo suporte técnico concedido.
REFERÊNCIAS BIBLIOGRÁFICA: [1] COTTON, F. A.; JAMERSON, J.D. 2,4,6-Tris(2-pyridyl)- 1,3,5-triazines Hydrolyze in the Presence of Copper(I1) to Form a Novel Bis(ary1)carboximidato Chelate Complex. Journal of the American Chemical Society, v. 98, p. 5397-5398, 1976.
[2] DIAS, V. L. N. et al. Electrochemical reduction of oxygen and hydrogen peroxide catalyzed by a surface copper(II)2,4,6-tris(2-piridil)-1,3,5-triazine complex adsorbed on a graphite electrode. Journal of Power Sources, v. 142, p. 10-17, 2005.
[3] JULVE, M. et al. Copper(II)-assisted hydrolysis of 2,4,6-tris(2-pyridyl)-1,3,5-triazine. Crystal structures of [bis(2-pyridylcarbonyl) amido] (pyridine-2-carboxamide) copper(II) trifluoromethanesulphonate and [bis(2-pyridylcarbonyl)amido] [2,4,6-tris(2-pyridyl)-1,3,5-triazine] copper(II) trifluoromethanesuiphonate. Journal of the Chemical Society Dalton, v. 9, p. 1681-1687, 1989.
[4] PAUL, P. et al. Synthesis and Characterization of Rhodium Complexes Containing 2,4,6-Tris(2-pyridyl)-1,3,5-triazine and Its Metal-Promoted Hydrolytic Products: Potential Uses of the New Complexes in Electrocatalytic Reduction of Carbon Dioxide. Inorganic Chemistry, v. 37, p. 5733-5742, 1998.
[5] HOLBREY, J. D. et al. The structure of [Co(H-tptz)Cl3]H2O (tptz=2,4,6-tri(2-pyridyl)-1,3,5-triazine) prepared by crystallization from the ionic liquid, N-butyl-N-methyl-pyrrolidinium
bis(trifluoromethanesulfonyl)imide. Journal of Chemical Crystallography, v. 36, n. 12, p. 799-804, 2006.
[6] FRISCH, M. J. et al. Gaussian 03, Revision B.04; Gaussian, Inc.: Pittsburgh PA, 2003.
[7] WEINHOLD, F.; FOSTER, J. P. Natural Hybrid Orbitals. Journal of the American Chemical Society, v. 102, p. 7211-7218, 1980.
[8] INGLEZ, S. D. et al. Tuning of Photochemical and Photophysical Properties of [RuII(2,2-bipyridine)2Lx] Complexes using Nonchromophoric Ligand Variations. Journal of the Brazilian Chemical Society, v. 21, n. 1, p. 157-68, 2010.
[9] BECKE, A. D., Density-functional Exchange-energy approximation with correct asymptotic behavior. Physical Review A, v. 38, p. 3098-3100, 1988.
[10] HUHEEY, J. E.; KEITER, E. A.; KEITER, R. L. Inorganic chemistry: principles structure and reactivity. 4. ed. New York: Haper Collins, 1993.
