ÁREA: Ensino de Química
TÍTULO: ATAQUE ELETROFILÍCO EM HALOBENZENOS: UMA ALTERNATIVA PARA O ENSINO UTILIZANDO O SOFTWARE SPARTAN
AUTORES: CARVALHO, R.B.F. (UFPI) ; ALMEIDA, A.A.C. (UFPI) ; SILVA, A.A.C.A. (UFPI) ; SANTOS, F.A. () ; LEAL, R.C (UFPI)
RESUMO: A química computacional consiste na aplicação dos métodos da mecânica quântica a problemas das diversas áreas da química. Em virtude da resolução de um grande número de equações necessário aos cálculos, a aplicação pragmática da mecânica quântica depende de programas computacionais específicos. Neste trabalho foi utilizado o software Pc Spartan PRO com a finalidade de prever teoricamente possíveis ataques eletrofílicos em reações de substituição no anel aromático mono-halogenado e em seguida comparar os resultados obtidos com os pressupostos da literatura. Os resultados fornecem aos alunos subsídios que facilitam a interpretação didática do fenômeno ocorrido.
PALAVRAS CHAVES: química computacional, química orgânica, substituição eletrofílica.
INTRODUÇÃO: A palavra aromática foi inicialmente utilizada para descrever algumas substâncias possuidoras de fragrâncias, entretanto, logo se observou que essas substâncias diferenciavam-se da maioria dos compostos orgânicos em relação ao seu comportamento químico (MCMURRY, 2005). Atualmente, utiliza-se o termo aromático para nos referir ao benzeno e seus derivados estruturais (AMARAL et al., 1980). Porém, devemos estar cientes que apenas os compostos que seguem a regra de Hückel (CAREY e SUNDBERG, 2007) são, de fato, verdadeiramente aromáticos.
Em uma substituição eletrofílica um composto aromático reage com o eletrófilo, onde normalmente este último substitui um dos átomos de hidrogênio do anel aromático, contudo pode haver outros casos (MCMURRY, 2005).
Os grupos substituintes no anel aromático são classificados e ordenados de acordo com a possibilidade de ativarem ou desativarem o anel em uma reação química (LEAL et al., 2010).
Em pesquisas químicas, um dos softwares de uso acadêmico que contempla o uso didático é o PC Spartan PRO, o mesmo possibilita a utilização de modelos moleculares na instrução da química e fornece aos estudantes métodos computacionais para resolução de problemas. O programa comporta diversas metodologias teóricas de cálculos, dentre elas podemos citar mecânica molecular, semi-empírico e Hartree-Fock (RAUPP et al., 2008).
O presente estudo tem como objetivo efetuar cálculos computacionais a fim de prever teoricamente possíveis ataques eletrofílicos em reações de substituição no anel aromático mono-halogenado e em seguida comparar os resultados obtidos com os pressupostos da literatura.
MATERIAL E MÉTODOS: Foi utilizado o programa computacional Pc Spartan Pro para o cálculo de geometria de equilíbrio em fase gasosa. Os métodos utilizados foram os semi-empíricos (AM1 e PM3) e o ab initio Hartree-Fock, com os conjuntos de base STO-3G, 3-21G(d) e 6-31G(d). As cargas atômicas foram obtidas para todos os métodos de cálculo com o objetivo de justificar o sítio ativo mais favorável nas reações de substituição eletrofílica. Os resultados foram interpretados e comparados com a abordagem discutida na literatura.
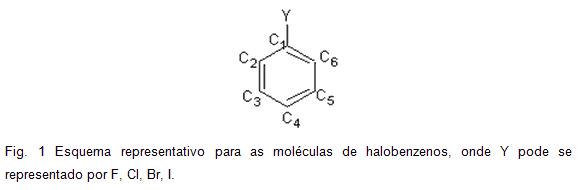
RESULTADOS E DISCUSSÃO: As moléculas dos halobenzenos (Fig. 1) possuem os halogênios como grupo desativante, porém influenciam um possível ataque eletrofílico em posições orto e para. Os efeitos e orientação dos seus substituintes estão fortemente relacionados com sua reatividade.
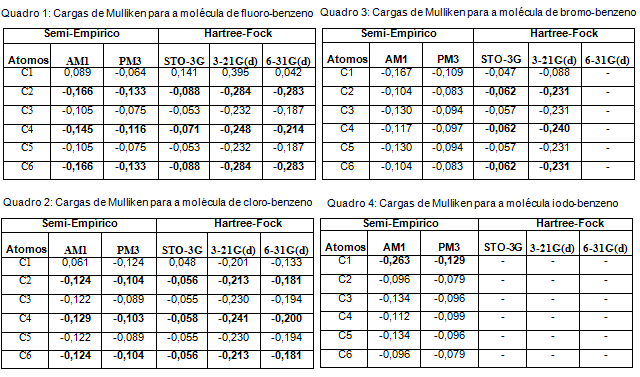
Os valores das cargas sob os átomos dos carbonos do anel aromático possibilitam a previsão de um possível ataque eletrofílico. Sendo assim, os resultados das diferentes metodologias computadas para os halobenzenos da Fig. 1 encontram-se expressos nos quadros 1, 2, 3 e 4.
Para as estruturas fluoro-benzeno (Quadro 1) e cloro-benzeno (Quadro 2) observa-se que a maior densidade eletrônica se encontra sob os átomos de carbono nas posições orto (C2 e C4) e para (C6), concordando com os resultados da literatura em relação a uma possível substituição eletrofílica em um anel aromático halogenado. De uma forma geral, verifica-se que o favorecimento da substituição eletrofílica na posição orto diminui à medida que aumenta o volume de Y (F, Cl, Br e I).
Para a estrutura do bromo-benzeno (Quadro 3) verifica-se que apenas os resultados obtidos com o método ab initio Hartree-Fock concordaram com a literatura, certamente devido a maior precisão do que a metodologia semi-empírica utilizada. O método Hartree-Fock com o conjunto de base 6-31G(d) não se adequa aos cálculos para a estrutura do bromo-benzeno, por isso não foram realizados cálculos para essas moléculas.
O método Hartree-Fock com os conjuntos de base STO-3G, 3-21G(d) e 6-31G(d) não foram realizados para a estrutura do iodo-benzeno devido a limitações da própria metodologia (Quadro 4). A metodologia semi-empírica não é concordante com a literatura, pois ambos os métodos AM1 e PM3 apontam para o C1 como o centro mais rico em densidade eletrônica.
CONCLUSÕES: Através dos métodos computacionais da Química Quântica é possível prever a posição mais provável para um ataque eletrofílico. Para as moléculas de fluoro-benzeno e cloro-benzeno a previsão da substituição concorda com a literatura. Na molécula de bromo-benzeno, apenas a metodologia Hartree-Fock apontou para as posições esperadas. Já a do iodo-benzeno foge a regra geral. Portanto, a utilização deste software pode servir como ferramenta mediadora do conhecimento na complementaridade de estudo de diversos fenômenos químicos ocorridos em nível atômico-molecular.
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA: AMARAL, L. F. P. et al. Fundamentos de Química Orgânica. São Paulo: Edgar Blücher, 1980.
CAREY, F. A. and SUMBERG, R. J., Advanced Organic Chemistry: Part A. 4rd ed. Plenum Press, New York, 2007.
LEAL, R. C.; LIMA, F. DAS C. A.; FEITOSA, C. M.; MOITA NETO, J. M. A Química Quântica na compreensão de teorias de Química Orgânica. Quim. Nova, Vol. 33, p. 1211-1215, 2010
MCMURRY, J. Química Orgânica, 6a ed. São Paulo: Pioneira Thomson Learning, v. 2, 2005.
RAUPP, D.; SERRANO, A.; MARTINS, T. L. C. A evolução da química computacional e sua contribuição para a educação em Química. Revista Liberato, Novo Hamburgo, v. 9, p. 13-22, 2008.
