
Realizado no Rio de Janeiro/RJ, de 14 a 18 de Outubro de 2013.
ISBN: 978-85-85905-06-4
ÁREA: Iniciação Científica
TÍTULO: Modelagem molecular de derivados bisquinolina com atividade antimalárica usando MEPs e orbitais de fronteiras
AUTORES: Rodrigues, E.C.R. (UNIFAP) ; Kawakami, M.Y.M. (UNIFAP) ; Cardoso, G.S.M. (UNIFAP) ; Lobato, C.C. (UNIFAP) ; Sousa, M.A.C. (IFAP) ; Hage Melim, L.I.S. (UNIFAP) ; Santos, C.B.R. (UNIFAP)
RESUMO: A malária é um problema de saúde pública que mais acometem seres humanos. A
resistência do protozoário aos medicamentos antimaláricos se destaca. Novos
antimaláricos e modificações de estruturas estão sendo propostos como forma de
tentar evitar ou minimizar essa resistência. O objetivo deste trabalho é estudar
a estabilidade química e a reatividade molecular dos derivados das quinolinas
com atividade antimalárica utilizando métodos de química quântica no nível de
teoria HF/3-21G*, HOMO, LUMO, GAP e MEP’s. O composto C4 apresentou um alto
valor de gap=-0.37336eV, enquanto que o composto C12 apresentou um menor valor
de gap=-0.38659eV, isso explica a razão desta molécula (C12) apresentar menor
atividade biológica, pois apresenta alta reatividade e menor estabilidade
molecular.
PALAVRAS CHAVES: bisquinolina; quinolina; antimaláricos
INTRODUÇÃO: A malária é um dos maiores problemas de saúde pública do mundo, já que é a
principal causa de perda econômica e alta morbidade e representa uma das doenças
infecciosas que mais acometem seres humanos. A quimioterapia da malária envolve
o uso de vários grupos de fármacos que compreendem a cloroquina, amodiaquina,
ferroquina, isoquina, pironaridina, naftoquina, aminoquinolizidina e
bisquinolinas (junção de duas 4-aminoquinolinas) (SOUZA, 2011).
Os vários esforços atuais têm se baseado na síntese de novas drogas e na
modificação molecular dos antimaláricos existentes, o que dificulta o
aparecimento de resistência, considerando que o processo de resistência a novos
fármacos, além de fisiologicamente complexo para o parasito, não é um processo
evolutivamente instantâneo. Por isso, em nenhuma outra área da química, o
conhecimento completo da estrutura molecular é tão essencial como na Química
Medicinal. Esta disciplina das Ciências Farmacêuticas estuda as origens
moleculares da atividade biológica dos fármacos, determinando os parâmetros que
relacionam estrutura e atividade e aplicando estes fundamentos no planejamento
racional dos fármacos (BARREIRO et, al, 1997).
Neste trabalho, foram utilizados métodos de química quântica no nível de teoria
HF/3-21G* para os novos derivados da bisquinolina com atividade antimalárica em
cepas D10 de plasmodium falciparum (HEERDEN et al, 2012), usando orbitais de
fronteiras (HOMO e LUMO), GAP como indicativo de reatividade química e
estabilidade molecular, e mapas de potencial eletrostático molecular (MEP's) que
foram avaliados e usados para identificar características chaves responsáveis
pela atividade destes novos derivados.
MATERIAL E MÉTODOS: O estudo de propriedades físico-químicas utilizando a química quântica é
utilizado para prever o comportamento químico de moléculas e, consequentemente,
a sua atividade biológica em diversas situações de estabilidade e reatividade
química (SANT’ANNA, 2009). As estruturas dos compostos selecionados foram
construídas com o programa Gauss View 1.0, de acordo com o seguinte
procedimento: inicialmente a estrutura do composto 1 foi construída e otimizada
com o método ab initio HF/3-21G*, e em seguida, os demais compostos foram
construídos a partir da mesma, então, obteve-se as estruturas otimizadas com
menor energia. A conformação mais estável de cada molécula estudada foi usada
para realizar os cálculos dos orbitais de fronteira (HOMO e LUMO) e mapas de
potencial eletrostático molecular (MEPs). As propriedades Químico-Quânticos
foram calculados usando o programa Gaussian 98 e HyperChem 6.02,
respectivamente. O potencial eletrostático foi obtido através de um conjunto de
cargas pontuais que representa o potencial quântico molecular de pontos
definidos em torno da molécula. Os mapas do potencial eletrostático molecular
representam uma superfície de contorno, e estes foram computados pelas cargas de
Millikan e pelas palavras chaves GFOLDPRINT POP=FULL. Os cálculos do orbital
HOMO (orbital molecular de maior energia) e do orbital LUMO (orbital molecular
de menor energia) assim como a visualização do Mapa de Potencial Eletrostático
Molecular, foram feitos a partir do software Molekel 4.2.
RESULTADOS E DISCUSSÃO: Os descritores químico-quânticos utilizados em estudos da relação estrutura
atividade relacionam-se à energia dos orbitais HOMO e LUMO. O orbital HOMO mede
o caráter elétron-doador de um composto, e o orbital LUMO mede o caráter
elétron-aceitador. Destas definições, observa-se que quanto maior a energia do
HOMO, maior a capacidade elétron-doadora, e quanto menor a energia do LUMO menor
será a resistência para aceitar elétrons. As energias do HOMO e do LUMO são
usadas como índices de reatividade química e são comumente correlacionadas com
outros índices, tais como afinidade eletrônica e potencial de ionização
(CARVALHO, 2003).
Na Figura 1 são mostrados os HOMO dos compostos estudados e podemos notar que
nesta região está ligada ao potencial de ionização do composto e caracteriza a
capacidade da molécula em realizar ataques nucleofílicos. A diferença entre as
energias dos orbitais HOMO-LUMO (gap) é um importante indicador de estabilidade
molecular. Moléculas com baixo valor de gap são geralmente reativas, enquanto
moléculas com alto valor de gap indicam alta estabilidade da molécula, no
sentido de baixa reatividade nas reações químicas (SANTOS et al, 2013).
gap = EHOMO – ELUMO
De acordo com a Tabela 1, o composto com maior atividade biológica é o C4, e o
de menor atividade é o C12. Essa diferença na atividade biológica deve-se ao
fato do composto C4 apresentar um alto valor de gap=-0, 37336. O composto C12
apresentou menor valor de gap=-0, 38659 ao comparar com as demais moléculas.
Isso explica a razão desta molécula apresentar menor atividade biológica, pois
apresenta alta reatividade e menor estabilidade molecular. Além disso, observou-
se menor valor de LUMO (0,07689eV) no composto C4 dentre as moléculas estudadas.
Figura 1
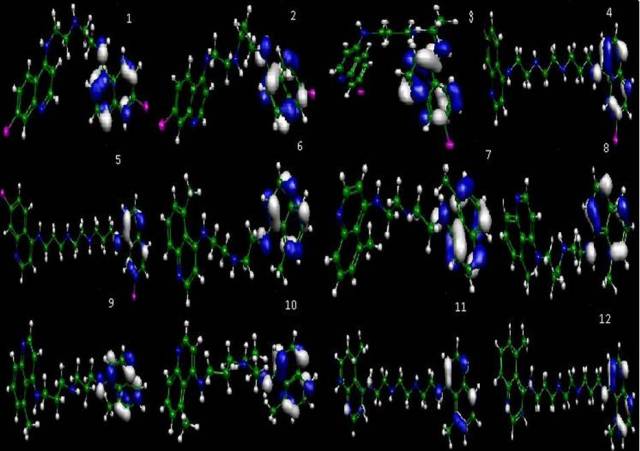
Orbital molecular ocupado de maior energia (HOMO)
dos compostos estudados com atividade antimalárica
usando o programa Molekel 4.2
Tabela 1
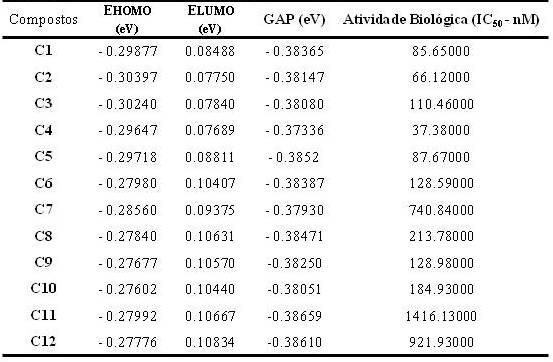
Descritores químico - quânticos utilizados para os
compostos estudados derivados de bisquinolina e
valores de IC50 experimental
CONCLUSÕES: De acordo com os dados obtidos, observou-se que os parâmetros analisados (HOMO,
LUMO, GAP e Mep’s) estão de acordo com os índices de atividade biológica (IC50) da
literatura, onde os compostos que tiveram um alto valor de GAP apresentaram
atividade antimalarica maiores dentre os compostos derivados da bisquinolina
estudados, o que foi mostrado pelo composto C4, que demonstrou ser o mais ativo e
o C12 que apresentou menor valor de GAP, sendo o menos ativo, ao comparar com os
demais valores de GAP das moléculas estudadas da série.
AGRADECIMENTOS: A Universidade Federal do Amapá (UNIFAP)
Ao LMQC(Laboratório de Modelagem Química Computacional)
REFERÊNCIAS BIBLIOGRÁFICA: BARREIRO, E. J., RODRIGUES, C. R. Modelagem molecular: uma ferramenta para o planejamento racional de fármacos em química medicinal. Rev. Química Nova. Rio de Janeiro - RJ, 1997.
CARVALHO, I. Introdução à modelagem molecular de fármacos no curso experimental de química farmacêutica. Quim. Nova, 2003
HEERDEN, L.V., CLOETE, T. T., BREYTENBACH, J. W., KOCK, C., SMITH, P. J., BREYTENBACH, J. C., N’DA, D. D. Synthesis and in vitro antimalarial activity of a series of bisquinoline and bispyrrolo[1,2a]quinoxaline compounds. European Journal of Medicinal Chemistry 55 (2012) 335-345.
SANT’ANNA, C. M. R. Métodos de modelagem molecular para estudo e planejamento de compostos bioativos: Uma introdução. Rev. Virtual Quim., 2009.
SANTOS, C. B. R., LOBATO, C. C., SOUSA, M. A. C., MACÊDO, W. J. C., CARVALHO, J. C. T. Reviews in Theoretical Science. No Prelo (2013)
SOUZA, N. B. Avaliação da atividade antiplasmodial de análogos da cloroquina. Juiz de Fora - MG, 2011.