
Realizado no Rio de Janeiro/RJ, de 14 a 18 de Outubro de 2013.
ISBN: 978-85-85905-06-4
ÁREA: Físico-Química
TÍTULO: Modelagem molecular de derivados de tiosemicarbazida com atividade antitripanosoma usando SAR e QSAR
AUTORES: Silva, M.C. (UNIFAP) ; Santos, S.S. (UNIFAP) ; Vieira, J.B. (UNIFAP) ; Lobato, C.C. (UNIFAP) ; Braga, F.S. (UNIFAP) ; Macêdo, W.J.C. (LACEN - PARÁ) ; Hage-melim, L.I.S. (UNIFAP) ; Santos, C.B.R. (UNIFAP)
RESUMO: Derivados de tiosemicarbazida são inibidores cruzaína e potentes agentes
antitripanosoma. Um conjunto de 15 derivados foi modelado usando métodos de
química quântica (HF/3-21G*) e análise estatística de regressão linear múltipla.
Os mapas de potencial eletrostático molecular foram computados pelas cargas de
Mullikan e comparados na região 1 (NH2), 2 (C) e 3,4 (N,N) da cadeia lateral. Os
descritores moleculares que apresentaram melhores correlações com a atividade
tripanosoma foram: Energia de hidratação, momento de dipolo total, coeficiente
de partição e LUMO. O modelo QSAR teve um grau de explicação de R=87.201%,
R2=76.040%, R2ajustado=62.349% da variabilidade dos valores observados para a
atividade. O teste de significância indicou valor de F=5.553 e erro padrão
estimado SEE=0.443.
PALAVRAS CHAVES: Modelagem molecular; atividade antitripanosoma; QSAR
INTRODUÇÃO: O desenvolvimento de novos agentes quimioterápicos contra infecções parasitárias
é uma das prioridades dos pesquisadores em todo o mundo (HERRLING, 2009). A
situação atual é composta por um conjunto limitado de drogas, incapazes de
fornecer soluções seguras e eficácias terapêuticas, sendo prejudicado por
aparecimento de cepas parasitários resistentes (RENSLO, 2006). A doença de
Chagas é causada pelo Trypanosoma cruzi e afeta cerca de 200 mil pessoas/ano,
dos quais cerca de 13 mil morrem de complicações da doença (DUSCHAK, 2007). As
proteases de T. cruzi são alvos interessantes para o desenvolvimento de novos
medicamentos, principalmente devido ao seu papel na replicação do parasita,
metabolismo e sobrevivência (O’BRIEN, 2008). Tiossemicarbazida é uma das
principais classes de inibidores de cruzaína, e foram descobertos por Du et al
(DU, 2002). Os compostos dessa classe têm mostrado atividade antitripanosoma
significativa e ativos em culturas de células infectadas com o T. cruzi. Neste
trabalho, foram utilizados métodos de química quântica no nível de teoria HF/3-
21G*, mapas de potencial eletrostático molecular (MEP's) que foram avaliados e
usados para identificar características chaves responsáveis pela atividade
destes novos derivados, orbitais de fronteiras (HOMO e LUMO) e construção de
modelo de calibração de relação estrutura– atividade quantitativa (QSAR) para um
conjunto de 15 novos derivados do tiossemicarbazida, baseando-se na construção
de modelos teóricos que descrevam a atividade de inibição cruzaína. Assim, este
estudo, contribui para a compreensão dos mecanismos moleculares envolvendo a
atividade antitripanosoma, visando o desenvolvimento de novos fármacos no
tratamento da doença de chagas.
MATERIAL E MÉTODOS: Um conjunto de 15 derivados tiosemicarbazidas (DU, 2002), com a atividade
antitripanosoma expresso com valores de concentração máxima inibitória (IC50)
foram selecionados, e os valores de IC50 foram convertidos no correspondente
pIC50 (-logIC50). Os compostos selecionadas foram construídos utilizando o
programa ChemSketch, e a pré-otimização molecular no programa HyperChem, pelo
método semi-empíricos. A parametrização com dados experimentais aumentou
significativamente a acuracidade química e a velocidade dos métodos de orbitais
moleculares simplificando o processo de cálculo. O sucesso desta abordagem é
indicado por inúmeros estudos mostrando dados energéticos que variam na faixa de
1,0 kcal/mol dos dados experimentais (BARREIRO et al., 1997). Após a pré-
otimização os compostos foram otimizadas com o método ab initio HF/3-21G* no
programa Gaussian 98, e dessa forma obteve- se as estruturas otimizadas com
menor energia. A partir da conformação mais estável foram realizados os cálculos
dos descritores: Energia total, energia eletrônica, entalpia (H), correção
térmica para a energia livre de Gibbs, soma dos elétrons, energia térmica livre,
momento de dipolo total (MD), GAP, moleza (S), dureza (n), eletronegatividade
(x), entropia (S), constante volume capacidade calorífica (CV), coeficiente de
partição (logP), volume molar (V), energia de hidratação (EH), refratividade,
sufarce (Approx), surface (Grid), usando o programa Gaussian 98 e HyperChem
6.02. Os MEP’s foram computados pelas cargas de Millikan usando o software
Molekel 4.2. Assim, como os orbitais de fronteiras HOMO (orbital molecular
ocupado de maior energia) e LUMO (orbital molecular desocupado de menor energia)
foram gerados e visualizados usando o mesmo software. A QSAR foi realizada
usando Statistica 6.1.
RESULTADOS E DISCUSSÃO: A energia do HOMO reflete o caráter elétron-doador do composto, e a energia do
LUMO reflete o caráter elétron-aceitador (CARVALHO, 2011). Na Figura 1 é
mostrado o LUMO dos compostos estudados, e podemos notar que nesta região está
ligada afinidade eletrônica, e caracteriza a suscetibilidade ao ataque por
nucleofilos. Por meio da diferença de energia entre o primeiro orbital virtual e
o último orbital ocupado, é possível determinar a quantidade de energia
necessária para a transição de um elétron do orbital HOMO para o orbital LUMO, o
chamado GAP de energia, ou banda proibida e define uma região energeticamente
proibida aos elétrons e indica a natureza do material (ALBUQUERQUE, 2008;
KIAMETIS, 2012). A construção do modelo QSAR foi baseada nas correlações de
descritores com o pIC50, e os descritores melhores correlacionados foram: EH,
MD, logP e LUMO. Na análise de correlação de Pearson entre os descritores e
pIC50, foi observado que a correlação entre os pares de descritores é maior que
-0.337552. Contudo, a correlação entre os descritores e pIC50 é maior que
-0.439691. Com os descritores correlacionados foi possível obter o modelo QSAR,
através da equação: pIC50=6.8185+0.067.(EH)+0.1278.(logP)+0.2154.(MD)-28.8262.
(LUMO), que apresentou um grau de explicação de R=87.201%, R2=76.040%,
R2ajustado=62.349% da variabilidade dos valores observados para a atividade. O
teste de significância indicou valor de F=5.553 e erro padrão estimado de
SEE=0.443. Na Tabela 1 é mostrada a atividade antitripanosoma (pIC50)
experimental e predito pela análise estatística de regressão linear múltipla e
resíduos para os compostos estudados.
Figura 1
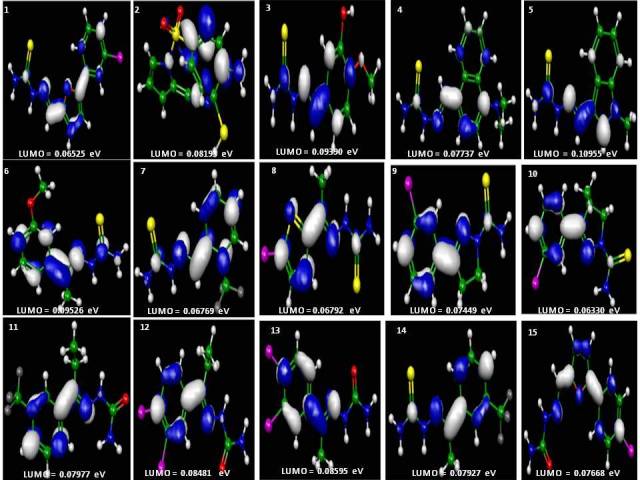
Orbital molecular desocupado de menor energia (LUMO)
dos compostos químicos com atividade antitripanosoma
usando o programa Molekel 4.2
Tabela 1
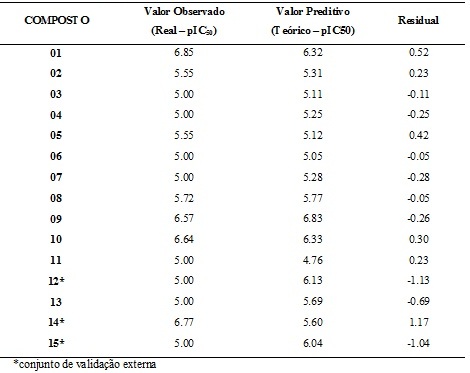
Atividade antitripanosoma (pIC50) experimental e
predito pela análise estatística de regressão linear
múltipla e resíduos para os compostos estudados.
CONCLUSÕES: A modelagem molecular é ferramenta inovadora para o planejamento de novos
fármacos. As propriedades moleculares EH, logP, MD e LUMO apresentaram alto
potencial de relevância para a atividade antitripanosoma, de acordo com os
parâmetros estatísticos obtidos pelo modelo QSAR. Os descritores selecionados
representam as características necessárias para quantificar a atividade
antitripanosoma de novos derivados de tiosemicarbazida, e isto significa um
resultado positivo, pois estas propriedades realizam importantes funções nas
estruturas dos compostos, estabilidade molecular e reatividade química.
AGRADECIMENTOS: Programa de Pós-Graduação da Rede BIONORTE
Laboratório de Modelagem e Química Computacional da Universidade Federal do Amapá
pelo apoio computacional.
REFERÊNCIAS BIBLIOGRÁFICA: ALBUQUERQUE, C. A. Modelagem Molecular Aplicada ao Desenvolvimento de Sistemas Nanoscopicos Bioativos. 2008. 133f. Dissertação (Mestrado em Ciências em Materiais para Engenharia) – Programa de Pós-Graduação em Materiais para Engenharia, Universidade Federal do Itajubá. Itajubá, MG. 2008.
BARREIRO, E. J.; RODRIGUES, C. R.; ALBUQUERQUE, M. G.; SANT’ANNA, C. M. R.; ALENCASTRO, R.B. Modelagem Molecular: Uma Ferramenta para o Planejamento Racional de Fármacos em Química Medicinal. Química Nova. v.20, n.1, p. 1-11. 1997.
CARVALHO, L. L. Modulagem Molecular de uma Série de Compostos Inibidores da Enzina Integrase do Virus HIV-1. 2011. 112f. Tese (Doutorado Físico-Química) – Instituto de Química de São Carlos, Universidade de São Paulo. São Paulo, SP. 2011.
DU, X. , GUO, C., E. HANSELL, P. S. DOYLE, C. R. CAFFREY, T. P. HOLLER, J. H. MCKERROW, F. E. COHEN, J. Med. Chem. 2002, 45, 2695.
DUSCHAK ,V. G., A. S. COUTO, Recent Pat. Antiinfect. Drug Discov. 2007, 2, 19.
HERRLING, P. L. Nat. Rev. Drug Discov. 2009, 8, 91.
KIAMENTIS, A. S. Modelagem Molecular de Potenciais Candidatos a Inibidores da Acetilcolinesterase. 2012. 90f. Tese (Doutorado) – Instituto de Física, Universidade de Brasília. Brasília, DF. 2012.
O’BRIEN, T. C., Z. B. MACKEY, R. D. FETTER, Y. CHOE, A. J. O’DONOGHUE, M. ZHOU, C. S. CRAIK, C. R. CAREY, J. H. MCKERROW, J. Biol. Chem. 2008, 283, 28934.
RENSLO, A. R., J. H. MCKERROW, Nat. Chem. Biol. 2006, 2, 701.