
Realizado no Rio de Janeiro/RJ, de 14 a 18 de Outubro de 2013.
ISBN: 978-85-85905-06-4
ÁREA: Físico-Química
TÍTULO: Estudo Teórico da reação de hidrogenação catalítica do CO2 com o complexo [RuPNPH2NO]
AUTORES: Spinelli, B. (UFMT) ; de Oliveira, L. (UFMT) ; da Silva, S. (UFMT) ; de Souza, G. (UFMT)
RESUMO: A reação de hidrogenação catalítica do CO2 utilizando como catalisador o
complexo metálico [Ru(PNP)H2NO], onde PNP= 2,6-bis(di-
isopropilfosfinometil)piridina, foi estudada computacionalmente utilizando a
teoria do funcional da densidade (DFT). Relatos recentes mostram que este
complexo com Irídio mostrou-se altamente eficiente na formação de ácido fórmico
como produto de reação, desta forma utilizou o metal Rutênio para tentar
aperfeiçoar a eficiência e introduzimos um ligante nitro para analisar o efeito
doador do mesmo grupo na reação. Desta forma, notamos que o efeito doador de
elétrons facilita a migração do hidrido, com uma Ea=9.269Kcal/mol, tendo um
∆Hr=-9.890Kcal/mol. A etapa de migração do próton ocorre com uma
Ea=6.367Kcal/mol, tendo um ∆Hr=+3.249Kcal/mol.
PALAVRAS CHAVES: Hidrogenação; CO2; DFT
INTRODUÇÃO: Com o desenvolvimento industrial, surgiram as ações decorrentes destas
atividades provocando alterações na biosfera, resultando na quase duplicação da
concentração dos gases de efeito estufa (GEE). Um dos principais GEE é o gás
carbônico (CO2), cujas emissões são provenientes da utilização de diversos tipos
de combustíveis. No mundo os países mais desenvolvidos são os maiores produtores
dos GEE, o Brasil ocupa a 21ª posição. Porém, ao incluir as queimadas e
desmatamentos, o Brasil passa a estar entre os seis primeiro emissores de GEE.
Tendo em vista que o aumento da liberação dos GEE é causado pela indústria que é
movida por combustíveis fósseis, petróleo, busca-se por novas fontes
alternativas de energia. Desta forma, é necessário que novas tecnologias sejam
estudas a fim de alterar este aspecto.
Apesar da utilização do CO2 em reações de síntese química contribuir
relativamente pouco para a mitigação da concentração do CO2 atmosférico, vários
outros fatores tornam-no um interessante reagente químico, como sua abundancia,
baixa toxicidade, baixo custo, e baixa temperatura crítica. Uma variedade de
produtos pode ser sintetizada utilizando CO2 como precursor, e entre elas a
produção de ácido fórmico tem ganhado muita atenção, devido ao fato de que o
ácido fórmico é um precursor de inúmeras reações orgânicas.
Reação de hidrogenação é a reação química entre hidrogênio molecular e um
elemento ou composto, geralmente na presença de um catalisador, reduzindo o
mesmo, para que cada átomo se ligue ao hidrogênio.
Diante deste contexto, a química computacional entrou em ação, tendo em vista
que tais reações devem ser estudadas em seus aspectos mecanisticos e
termodinâmicos, logo, a área computacional proporciona uma ferramenta muito útil
para esta finalidade.
MATERIAL E MÉTODOS: Todos os cálculos computacionais foram realizados utilizando o programa
Gaussian09, utilizando a teoria do funcional da densidade (DFT), sendo o método
B3LYP e o conjunto de funções de base DGDZVP, foi utilziado como método de
solvatação, IEFPCM, utilizando como solvente a água. Todos os cálculos foram
visualizados utilizando o programa GaussView 5.0.
O complexo inicial foi otimizado previamente, e então se obteve as energias
livre e entalpia corrigidas, logo após, foi introduzido o CO2 no sistema,
realizando uma otimização e varredura na ligação a ser analisada, transferência
do hidrído, logo após o produto desta varredura foi otimizado e então se
calculou o QST3, a fim de conhecer o estado de transição da reação de migração
do hidrído, após foi realizado a isomerização do o íon formil interagindo com o
rutênio via hidrogênio, Ru-H-CO2, para que este se ligue ao núcleo metálico pela
ligação Ru-O-CHO, e então o produto foi otimizado e calculado o QST3 a fim de
conhecer o estado de transição da reação de isomerização. Após esta etapa, foi
feita a adição de uma molécula de H2, que foi introduzida no complexo, forçando
a saída do íon formil, sendo feita via otimização e varredura, logo após, foi
realizado o QST3 da reação de inserção da molécula de dihidrogênio a fim de se
conhecer o estado de transição da reação. Então foi realizado outra varredura
com o produto do sistema da inserção otimizado, e fazendo a varredura da
migração do próton da molécula de dihidrogênio para o íon formil, formando o
ácido fórmico. Foi feito após a varredura o QST3, a fim de conhecer o estado de
transição da migração do próton. Então calculou-se todos as energias do sistema.
RESULTADOS E DISCUSSÃO: Foi realizado os cálculos do ciclo catalítico da hidrogenação do CO2, formando
um ciclo de 4 etapas, sendo os 4 estados de transição do ciclo, onde o TS1,2
representa a migração do hidrido para o CO2, o TS2,3 representa a isomerização
do íon formil, o TS3,4 representa a coordenação da molécula de dihidrogênio, e o
TS4,5 representa a transferência do próton.
Segue na figura 1 o ciclo catalítico e na figura 2 os 4 estados de transição do
ciclo, sendo que nesta figura os grupos isopropil foram omitido para melhor
visualização.
Desta forma obtivemos um ciclo catalítico com valores de energia para cada etapa
da reação, sendo que na primeira etapa, migração do hidrido, esta ocorre com uma
barreira de ativação de +9.269Kcal/mol e com um ∆Hr de -4.421Kcal/mol. A segunda
etapa, isomerização do formil, ocorre com uma energia de ativação de
+1.543Kcal/mol e com um ∆Hr de -8.511Kcal/mol. A terceira etapa, inserção do
hidrogênio molecular no núcleo do complexo, ocorre com um ∆Gr de +17.913kcal/mol
e com um ∆Hr de +25.146Kcal/mol sendo a etapa com maior dificuldade de
ocorrência, devido à grande estabilidade do produto isomerizado.
A quarta etapa, clivagem da ligação H2 e migração do próton para o íon formil,
formando o produto ácido fórmico, ocorre com uma barreira de ativação de
+6.367Kcal/mol e com um ∆Hr de +3.249Kcal/mol. Após foi feito a análise da
retirada do produto da reação para se verificar os valores de variação de
energia de Gibbs e entalpia, sendo este, considerado os valores globais para a
reação, sendo o ∆Hg de +3.178Kcal/mol e ∆Hg de -5.120Kcal/mol.
Todos os produtos e reagentes foram otimizados, as energias de ativação,
variação da energia livre da reação e variação da entalpia de reação foram
calculados.
Figura 1
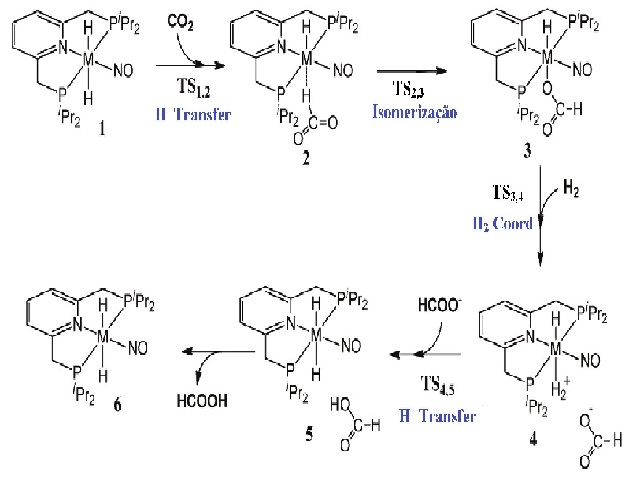
Ciclo catalitico completo da reação
Figura 2
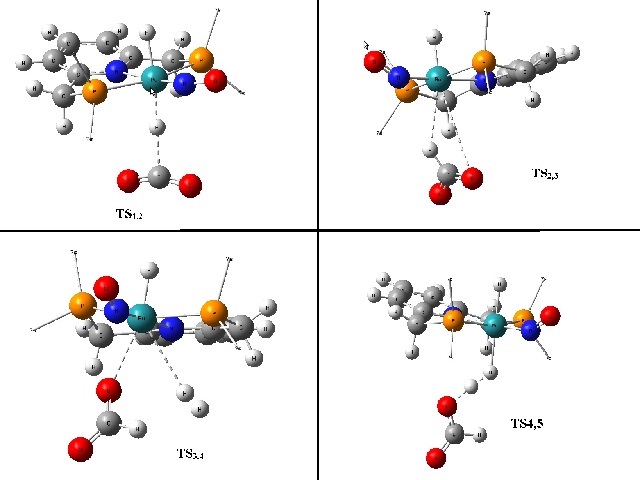
Esquema ilustrando os quatro estados de transição do
ciclo.
CONCLUSÕES: Podemos concluir que com a utilização do grupo nitro com um ligante do complexo
metálico, este proporciona um efeito doador de elétrons para o núcleo, que
facilita a primeira etapa da reação, a migração do hidrido para o CO2, facilita a
isomerização, tendo uma baixa energia de ativação, tendo um produto estável,
complexo com formil isomerizado, desta forma, as seguintes etapas da reação se
mostram muito desfavoráveis. Apesar da reação possuir um valor positivo para a
variação de energia livre, +3.178, notamos um word ∆H de -5.120, sendo esta reação
levemente exotérmica.
AGRADECIMENTOS: CAPES,CNPq, FINEP e FAPEMAT
REFERÊNCIAS BIBLIOGRÁFICA: Carey F. A.; Sundberg R. J. 2007. Advanced Organic Chemestry, Part A: Structure and Mechanisms. Ed. Springer, 5th ed.
Morgon N. H.; Coutinho K. 2007. Métodos de Química Teórica e Modelagem Molecular. São Paulo/SP, Ed. Livraria da Física.
Rocha, M. T. 2003. Aquecimento global e o mercado de carbono: Uma aplicação do modelo cert. Doutorado em agronomia, USP/ Sorocaba-SP. Dissertação.
Sakaki, S.; Musashi, Y. 2002. Hydrogenation of Carbon Dioxide. Kluwer Academic Publishers. P. 79-105.
Tomberg, A. 2009. Gaussian 09W Tutorial, an introduction to computational chemistry using G09W and Avogadro software.
Yang, X. 2001. Hydrogenation of Carbon Dioxide Catalyzed by PNP Pincer Iridium, Iron, and Cobalt Complexes: A Computational Design of Base Metal Catalysts Am. Chem. Soc. Catal. 1: 849-854.