SÍNTESE DE AMIDAS UTILIZANDO ADUTOS DE MORITA-BAYLIS-HILMAN
ISBN 978-85-85905-23-1
Área
Química Orgânica
Autores
Marques, S.D.G. (UFPB) ; Antunes, M.S.M. (UFPB) ; Mosquera, A.M. (USC) ; Souza, M.F.V. (UFPB) ; Luna-freire, K.R. (UFPB)
Resumo
As amidas são uma classe importante de substâncias orgânicas encontrada em abundância na natureza como em proteínas, em polímeros biológicos e sintéticos. As funções amida não estão limitadas a sistemas biológicos, mas também em uma enorme variedade de moléculas sintéticas, incluindo diversos medicamentos comercializados. A fim de sintetizar amidas, avaliando métodos que possibilitem obter um maior grau de pureza nas reações, foram utilizados adutos de MBH (Morita-Baylis-Hillman) como substratos para a síntese destas moléculas, tendo como reagentes de partida aldeídos aromáticos e aminas comerciais, em uma síntese que segue de 3-5 etapas reacionais. Por meio deste trabalho foi possível sintetizar 40 moléculas, sendo 20 amidas, destas 18 são inéditas na literatura.
Palavras chaves
Síntese; Adutos de MBH; Amidas
Introdução
As amidas são substâncias orgânicas derivados de ácidos carboxílicos pela substituição da hidroxila (‒OH) pelo grupamento amino (‒NH2). De acordo com a substituição dos hidrogênios, as amidas podem ser classificadas como monossubstituídas ou dissubstituídas(USBERCO,2006). As amidas são uma classe importante de substâncias orgânicas encontrada em abundância na natureza como em proteínas, em polímeros biológicos e sintéticos. Além de estarem presentes em sistemas biológicos, as funções amida também se apresentam em uma enorme variedade de moléculas sintéticas, incluindo diversos medicamentos comercializados (KUNG et al., 2010) (MACIEL, 2016)(CAVALCANTE, 2010) (GLOMB & PFAHLER, 2001). A síntese de amidas pode ser realizada a partir de adutos de Morita-Baylis- Hillman (MBH) que tem se mostrado eficiente no que se refere à preparação de várias moléculas comercialmente úteis, de maneira simples e versátil (COELHO & ALMEIDA, 2000)(BASAVAIAH, & NAGANABOINA, 2018). A reação de MBH (Figura 01), conhecida desde 1968 (DA CRUZ et al., 2005), pode ser definida, de um modo geral, como uma reação de condensação entre carbonos eletrofílicos sp2 (geralmente um aldeído) e a posição α de uma olefina contendo grupos retiradores de elétrons (derivado acrílico), catalisada por amina terciária ou fosfina, levando à formação de uma nova ligação σ C-C. A reação de MBH pode em alguns aspectos ser semelhante a algumas metodologias para obtenção de substâncias β-hidroxicarboniladas. No entanto, esta reação apresenta grande potencial por apresentar características fundamentais para a eficiência de um método sintético, como ser regio e quimiosseletiva, além de ser econômica e atuar em condições reacionais brandas providenciando moléculas polifuncionalizadas, através de sucessivas interconversões de grupos funcionais, podendo, desta forma, permitir o acesso a importantes intermediários sintéticos (COELHO & ALMEIDA, 2000) (BASAVAIAH, & NAGANABOINA, 2018). Envolvendo uma economia de átomos, onde todos os átomos presentes nos reagentes de partida estão incorporados no produto, a reação de MBH se apresenta como muito útil e versátil, pois combina reações de aldol e Michael em um único passo, fornecendo um metileno hidroxicarbonilo ou metileno aminocarbonilo organocatalisado (CARRASCO-SANCHEZ et al., 2009). Estes compostos são de construção multifuncional, sendo utilizados blocos na síntese de produtos naturais e compostos farmacêuticos relevantes para a saúde (DAS et al., 2006) (AMARANTE et al., 2008) (WASNAIRE et al., 2006) (LUNA-FREIRE et al., 2014) (LUNA-FREIRE et al., 2011). Grupos de pesquisas vêm utilizando os adutos de MBH como substratos para a síntese de moléculas biologicamente ativas (TRAZZI et al., 2010; SREEVANI et al., 2011; AMARANTE et al., 2010; KAUR et al., 2011). Estes substratos apresentam como principal característica a presença de três grupos funcionais: um grupo hidroxila, uma olefina e um éster, cetona, nitrila, sulfona ou fosfonato (COELHO & ALMEIDA, 2000). Na síntese de amidas os agentes acopladores são frequentemente utilizados. Dentre estes, os que já incorporam o grupo fenol estão sendo usados como catalisadores eficientes, estando também, em sua maioria, comercialmente disponíveis. Eles podem ser classificados de acordo com sua natureza, isto é: sais de urônio, fosfônio e imonium. Dentre os sais de fosfônio, o Benzotriazol-1-il-oxi-tris-(dimetilamino)-fosfonium hexafluorofosfato (BOP), também chamado de reagente de Castro (CASTRO et al., 1975), é o primeiro exemplo publicado de reagente a base do sal de onium hidroxibenzotriazol (HOBt). Para a formação da amida o acoplamento é realizado misturando-se o ácido desejado a amina na presença de BOP e trietilamina ou base de Hunig. O ácido desprotonado primeiro reage com BOP para gerar um acilfosfônio ativado e o HOBt. O HOBt prontamente reage com o ácido ativado para produzir um éster Bt reativo, que finalmente sofre aminólise. A força motriz desta reação baseada em fosfônio é gerar o correspondente óxido (CASTRO et al., 1976) (MONTALBETTI & FALQUE, 2005). Destarte, tem-se como objetivo a síntese de amidas, avaliando métodos que possibilitem obter um maior grau de pureza nas reações, possibilitando o desenvolvimento de moléculas bioativas utilizando poucas etapas reacionais e reações de fácil execução.
Material e métodos
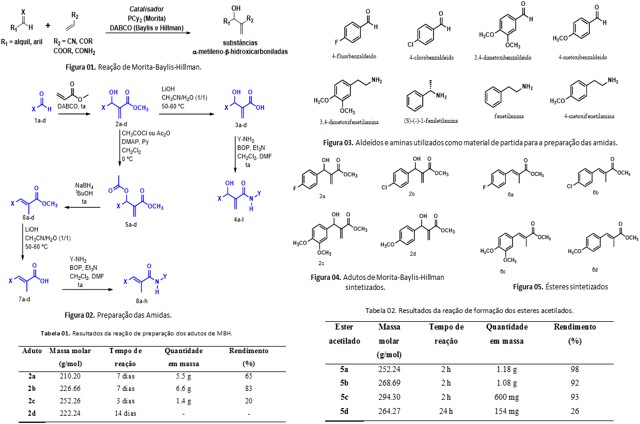
O esquema das etapas de formação das amidas segue na Figura 02. Os aldeídos (Figura 03) (1 equiv.) foram submetidos a reação de MBH , onde ocorreu a condensação do aldeído com o acrilato de metila (10 equiv.), sendo catalisada pela amina terciária DABCO (1,4-diazabiciclo[2.2.2]octano) (1 equiv.). As reações foram realizadas sob agitação magnética até o consumo do material de partida. O excesso de acrilato de metila foi evaporado a pressão reduzida obtendo um resíduo que foi extraído com água (1x) e, em seguida com solução saturada de NaCl (2-3x). Após a formação dos adutos de MBH, parte de cada aduto foi submetida a uma reação de hidrólise e outra parte a uma reação de acetilação (Figura 02). Para a acetilação do grupo hidroxila dos adutos de MBH, a uma solução deste (1 equiv) em diclorometano (CH2Cl2), foi adicionado piridina (2 equiv.) quantidade catalítica de 4-dimetilaminopiridina (DMAP) e cloreto de acetila (5a,b e c) ou anidrido acético (5d) (4 equiv.), em banho de gelo. A mistura foi mantida sob agitação magnética à temperatura ambiente até o consumo do material de partida. Em seguida, evaporou-se a piridina e o CH2Cl2 em rotaevaporador. A mistura reacional foi diluida em AcOEt e a fase orgânica foi lavada em solução de ácido acético à 10% (2x) e solução de NaHCO3 (2x) (STORK et al., 1978). A partir dos ésteres obtidos, seguiu-se para a etapa de formação dos ácidos por hidrólise em meio básico. Para a realização dessa reação foi utilizado o ester (1 equiv.), LiOH ( 10 equiv.) e acetonitrila/água (1/1). A reação ficou sob agitação magnética e em aquecimento 50-60ºC. Terminada a reação, a acetonitrila foi evaporada, o resíduo solubilizado em acetato de etila e submetido a extração com água e HCl 25% (AMARANTE et al., 2011). Para realizar a última etapa no processo de preparação das amidas, foram utilizados como material de partida os ácidos obtidos nas reações anteriores e as aminas obtidas comercialmente (Figura 03). Em um balão mantido sob agitação magnética, o ácido orgânico (1 equiv.) foi dissolvido em DMF e em trietilamina (Et3N) (1 equiv.). A solução foi arrefecida em um banho de gelo (0ºC) e a amina adicionada. Na sequência uma solução de BOP (1 equiv.) em CH2Cl2 foi adicionado ao balão. A reação foi agitada a 0ºC durante 30 min e depois por um período adicional à temperatura ambiente durante 3 horas. Terminada a reação, o CH2Cl2 foi removido sob pressão reduzida e o produto foi extraído com AcOEt (3x) e água. A fase orgânica foi lavada sequencialmente com HCl a 1N, água, NaHCO3 a 1M e água novamente (FU et al. 2010). Em todas as reações a fase orgânica foi seca com Na2SO4, filtrada e concentrada em rotavapor, sendo os produtos purificados por cromatografia em uma coluna de sílica gel, usando Hex:AcOEt como fase móvel.
Resultado e discussão
Inicialmente foi realizado a síntese dos adutos de Morita-Baylis-Hillman
(MBH) a partir de aldeídos aromáticos comerciais, utilizando o acrilato de
metila em excesso, como aceptor de Michael (Figura 02). As reações foram
monitoradas por CCDA (cromatografia em camada delgada analítica), a fim de
controlar o tempo reacional por meio do consumo do aldeído. Vale ressaltar
que em todas as reações, não houve a formação de coprodutos, sendo
necessário uma purificação por cromatografia de coluna em sílica flash para
separar o aldeido do aduto de MBH formado.
Pode-se observar pela Tabela 01 que a condição experimental utilizada não
gerou resultados muito satisfatórios, considerando tempo reacional e/ou
rendimentos, principalmente para os adutos 2c e 2d. Estes adutos apresentam
em sua estrutura o grupo metoxila que aumenta a densidade eletrônica na
carbonila do aldeído, o que torna a reação mais lenta, com rendimentos de
moderados a baixos.
Não foi possível calcular o rendimento da reação de formação do aduto 2d
pois não foi possível realizar a sua purificação com eficiência, sendo o
mesmo posto para reagir as etapas seguintes ainda impuro com o aldeído de
partida.
Diante do exposto, em relação à reação de MBH, sabe-se que esta apresenta
como desvantagem a velocidade de reação, contudo optou-se por obter a
quantidade máxima desejável do produto devido às condições laboratoriais.
Após o isolamento por cromatografia em coluna os adutos foram submetidos a
técnicas espectroscópicas de RMN1H e/ou RMN13C.
Para a obtenção de mais derivados de MBH, foi realizado a acetilação dos
adutos, utilizando como agente acetilante o anidrido acético ou o cloreto de
acetila, e a piridina (base fraca) como catalisador.
Através dos dados observados na Tabela 02 é possível destacar que as reações
de formação dos ésteres acetilados 5a, b e c, foram satisfatórias, levando
em consideração o tempo reacional e o rendimento apresentado. Porém a reação
de formação do éster 5d teve seu rendimento comprometido principalmente pelo
fato do reagente de partida, o aduto MBH, não está puro. Além disso, foi
utilizado para a formação deste éster o anidrido acético que reage mais
lentamente quando comparado ao cloreto de acetila.
Seguidamente foi realizada a desacetilação dos adutos com a remoção da
hidroxila acetilada, utilizando o NaBH4 como agente redutor. A reatividade
deste reagente está diretamente ligada a diferença de eletronegatividade
entre o hidrogênio e o átomo central do ânion (B-). O borohidreto de sódio é
relativamente insolúvel em solventes etéreos (0,1g/100g de THF, 0.8g/100g de
1,2-dimetoxietano), e como a água e álcoois em meio básico ou neutro reagem
bem lentamente com o NaBH4, o terc-BuOH foi escolhido como solvente (COSTA
et al., 2003).
Ao final da reação, os produtos foram purificados por coluna cromatográfica,
onde foram obtidos os ésteres 6a, b, c, e d (Figura 05).
A formação dos ácidos se deu por hidrólise em meio básico, a partir dos
ésteres obtidos nas reações anteriores. A denominação de hidrólise é dada
para reações que envolvam a quebra de uma molécula por ação da água, onde
ocorre uma alteração de íons, sendo liberados para a solução cátions ou
ânions. Dessa forma, determinada molécula fragmenta-se e tem suas ligações
complementadas com os íons resultantes da molécula de água. Na maioria
destas reações, para que ocorra uma hidrólise rápida e completa, é
necessária a utilização de altas pressões e altas temperaturas e agentes
catalisadores (PERUZZO & CANTO, 2006).
Durante a extração do produto formado, que é o sal resultante da reação, foi
necessário uma solução de HCl em concentração suficiente (25%) para a
formação dos ácidos desejados (Figura 06). Estes foram purificados em coluna
cromatográfica. Observa-se, na Tabela 03, rendimentos de moderados a altos,
em baixos tempos reacionais (1-2h).
As amidas foram obtidas a partir da reação dos ácidos adquiridos nas etapas
anteriores (Figura 06), e aminas obtidas comercialmente (Figura 03),
utilizando dimetilformamida (DMF), trietilamina (Et3N), e agente de
acoplamento BOP, sendo a reação monitorada por CCDA, utilizando um sistema
Hex:AcOEt, segundo a metodologia de FU e colaboradores (2010).
Nessa reação de aminólise, a Et3N tem o papel de ionizar o ácido para que
ocorra o ataque da amina que é um reagente nucleófilo forte. O banho de gelo
utilizado nos primeiros 30 minutos da reação é necessário para impedir o
ataque de outros nucleófilos, já que a amina, mesmo em baixas temperaturas,
ainda permaneça como um bom nucleófilo (KIM & PATEL, 1994).
De acordo com Tabela 04, pode-se observar que as reações apresentaram bons
rendimentos entre 71-99%, sendo a amida 4a com maior rendimento e a amida 4d
a de menor rendimento. No entanto, provavelmente devido a problemas durante
a purificação nas etapas anteriores, não foi possível purificar as amidas
4i, 4j, 4l, 4m e 8h e, portanto, calcular os rendimentos destas reações.
Foram preparadas vinte amidas (Figura 07) as quais foram identificados por
métodos espectroscópicos de infravermelho, RMN de 1H, 13C uni e/ou
bidimensional
As amidas 4b e 4c, assim como as amidas 4e e 4f (Figura 07), são produtos
provenientes de uma mesma reação, onde foi possível a separação por coluna
cromatográfica por apresentarem uma leve diferença de polaridade. Na reação
das amidas 4b e 4c, foi purificado 150 mg da amida 4b e 50 mg da amida 4c,
além de 100 mg da mistura das duas amidas. Por sua vez, na reação das amidas
4e e 4f, foi obtido 102 mg da amida 4e e 63 mg da amida 4f, e 73 mg da
mistura das duas amidas.
Ao analisar os espectros de RMN uni e bidimensionais das amidas 4b e 4c, não
foram observadas diferenças que levassem a identificação dos
estereoisômeros, o mesmo aconteceu com as amidas 4e e 4f, não sendo possível
identifica-las por estes métodos. Foi realizado o experimento de desvio da
luz polarizada de cada amida, sendo calculado o seu poder rotatório (Tabela
05), confirmando que estas moléculas, opticamente ativas, são diferentes
diastereoisômeros.
A reação de formação da amida 4g também forma dois estereoisômeros, porém,
diferente das misturas anteriores não foi possível separar estes
diastereoisômeros por cromatografia em coluna, podendo ser verificado no
espectro RMN1H e RMN13C sinais duplicados e integrais para mais de uma
amida.
Figuras 01, 02, 03, 04 e 05. Tabelas 01 e 02
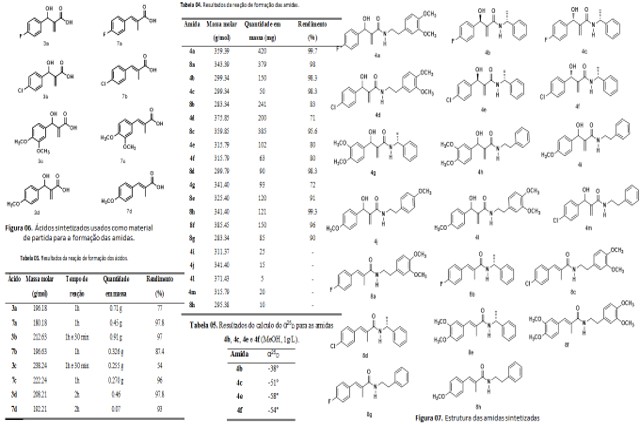
Figura 06 e 07. Tabelas 03, 04 e 05
Conclusões
Por meio deste estudo foi possível realizar: - A síntese de quatro adutos (2a-d) de MBH em reações com média de rendimento de 56%; - A preparação de três ésteres acetilados (5a-c) com média de rendimento de 94% usando o cloreto de acetila e a preparação de um éster (5d) usando anidrido acético. Estes foram seguidamente desacetilados formando os ésteres 6a-d; - A formação de oito ácidos (3a-d e 7a-d) em reações com média de rendimento de 87%; - E a obtenção de vinte amidas através de reações de fácil execução com média de rendimento de 90%. Com exceção das amidas 8c e 8f, todas as demais amidas são inéditas; - Foi possível obter uma média de rendimento de 82% incluindo todas etapas reacionais de formação das amidas. Destarte, foram preparadas um total de 40 moléculas, estas moléculas foram elucidadas por meio de técnicas espectroscópicas de RMN 1H e 13C e/ou IV, como também, em alguns casos, foi verificado o poder rotatório por polarimetria.
Agradecimentos
A CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) e ao CNPQ (Conselho Nacional de Desenvolvimento Científico e Tecnológico) pelo apoio financeiro.
Referências
AMARANTE, G.W.; REZENDE, P.; CAVALLARO, M.; COELHO, F. Acyloins from Morita−Baylis−Hillman adducts: an alternative approach to the racemic total synthesis of bupropion. Tetrahedron Letters, n. 49, p. 3744–3748, 2008.
AMARANTE, G. W.; CAVALLARO, M.; COELHO, F. Highly diastereoselective total synthesis of the anti-tumoral agent (+/-)-Spisulosine (ES285) from a Morita-Baylis-Hillman adduct. Tetrahedron Letters, v. 51, p. 2597, 2010.
AMARANTE, G. W.; CAVALLARO, M.; COELHO, F.; Hyphenating the Curtius Rearrangement with Morita-Baylis-Hillman Adducts: Synthesis of Biologically Active Acyloins and Vicinal Aminoalcohols. Journal of the Brazilian Chemical Society, v. 22, n. 8, p. 1568-1584, 2011.
BASAVAIAH, D.; NAGANABOINA, R. T. The Baylis–Hillman reaction: a new continent in organic chemistry – our philosophy, vision and over three decades of research. New Journal of Chemistry, Advance Article, p. 1-3, 2018.
CARRASCO-SANCHEZ, V.; SIMIRGIOTIS, M. J.; SANTOS, L. S. The Morita-Baylis-Hillman Reaction: Insights into Asymmetry and Reaction Mechanisms by Electrospray Ionization Mass Spectrometry. Molecules, v. 14, p. 3989-4021, 2009.
CASTRO, B.; DORMOY, J. R.; EVIN, G.; SELVE, C. Reactifs de couplage peptidique I (1) - l'hexafluorophosphate de benzotriazolyl N-oxytrisdimethylamino phosphonium (B.O.P.). Tetrahedron Letters, n. 14, p. 1219–1222, 1975.
CASTRO, B.; DORMOY, J.-R.; DOURTOGLOU, B.; EVIN, G.; SELVE, C.; Ziegler, J.-C. Peptide Coupling Reagents VI1. A Novel, Cheaper Preparation of benzotriazolyloxytris [dimethylamino] phosphonium hexafluorophosphate (BOP Reagent). Synthesis, p. 751-752 , 1976.
CAVALCANTE, J.M.S.; NOGUEIRA, T.B.S.S.; TOMAZ, A.C.A.; SILVA, D.A.; AGRA, M.F.; SOUZA, M.F.V.; CARVALHO, P.R.C.; RAMOS, S.R.; NASCIMENTO, S.C.; GONÇALVES-SILVA, T. Steroidal and phenolic compounds from Sidastrum paniculatum (L.) Fryxell and evaluation of cytotoxic and anti-inflammatory activities. Química. Nova, v. 33, n. 4, p. 846-849, 2010.
COELHO, F.; ALMEIDA, W. P. Reação de Baylis-Hillman: uma estratégia para a preparação de intermediários multifuncionalizados para síntese orgânica. Química Nova, v. 23, n.1, 2000.
COSTA, P. R. R.; PILLI, R. A.; PINHEIRO, S.; VASCONCELLOS, M. L. A. A. Substâncias carboniladas e derivados. Porto Alegre: Bookman, p.180, 2003.
DA-CRUZ, A.; PIRMEZ, C. In: Dinâmica das doenças infecciosas e parasitárias, Editor José Rodrigues Coura – Rio de Janeiro, Guanabara Koogan, p. 697-712, 2005.
DAS, B.; CHOWDHURY, N.; BANERJEE, J.; ANJOY, M. A facile one-pot stereoselective synthesis of trisubstituted (E)-2-methylalk-2-enoic acids from unactivated Baylis−Hillman adducts and a simple access to some important insect pheromones. Tetrahedron Letters, n. 47, p. 6615–6618, 2006.
FU, J.; CHENG, K.; ZHANG, Z.; FANG, R.; ZHU, H. Synthesis, structure and structure–activity relationship analysis of caffeic acid amides as potential antimicrobials. European Journal of Medicinal Chemistry, v. 45: p. 2638–2643, 2010.
GLOMB, M. A.; PFAHLER, C. Amides Are Novel Protein Modifications Formed by Physiological Sugars. Journal of Biological Chemistry, v. 276, p. 41638–41647, 2001
KAUR, J.; KUMAR, P.; TYAGI, S.; PATHAK, R.; BATRA, S.; SINGH, P.; SINGH, N. In silico screening, structure-activity relationship, and biologic evaluation of selective pteridine reductase inhibitors targeting visceral leishmaniasis. Antimicrob. Agents Chemother, v. 55, n. 2, p. 659-66, 2011.
KUNG, P. P.; HUANG, B. W.; ZHANG, G.; ZHOU, J. Z.; WANG, J.; DIGITS, J. A.; SKAPTASON, J.; YAMAZAKI, S.; NEUL, D.; ZIENTEK, M.; ELLERAAS, J.; MEHTA, P.; YIN, M. J.; HICKEY, M. J.; GAJIWALA, K. S.; RODGERS, C.; DAVIES, J. F.; GEHRING, M. R. Dihydroxyphenylisoindoline Amides as Orally Bioavailable Inhibitors of the Heat Shock Protein 90 (Hsp90) Molecular Chaperone. Journal of Medicinal Chemistry. v. 53, p. 499–503, 2010.
LUNA-FREIRE, K. R.; TORMENA, C.; COELHO, F. Heck Reaction on Morita-Baylis-Hillman Adducts: Diastereoselective Synthesis of Pyrrolizidinones and Pyrrolizidines. Synlett (Stuttgart), v. 2011, p. 2059-2063, 2011.
LUNA-FREIRE, K. R.; S.; JOÃO PAULO S.; RESENDE, J. A.L.C.; TORMENA, C. F.; OLIVEIRA, F. L.; APARICIO, R.; COELHO, F. An asymmetric substrate-controlled Morita-Baylis-Hillman reaction as approach for the synthesis of pyrrolizidinones and pyrrolizidines. Tetrahedron, v. 70, p. 3319-3326, 2014.
MACIEL, J. K. S.; CHAVES, O. S.; BRITO FILHO, S. G.; TELES, Y. C. F.; FERNANDES, M. G.; ASSIS, T. S.; FERNANDES, P. D.; ANDRADE, A. P.; FELIX, L. P.; SILVA, T. M. S.; RAMOS, N. S. M.; SILVA, G. R.; SOUZA, M. F. V. New Alcamide and Anti-oxidant Activity of Pilosocereus gounellei A. Weber ex K. Schum. Bly. ex Rowl. (Cactaceae). Molecules, v. 21, 2016.
MONTALBETTI, C. A. G. N.; FALQUE, V. Amide bond formation and peptide coupling. Tetrahedron, v. 61, p. 10827–10852, 2005.
PERUZZO, F. M. (Tito).; CANTO, E. L. Química na Abordagem do Cotidiano. São Paulo: Moderna, v.1, 2006.
SREEVANI, R.; MANJULA, A.; RAO, B. V. J. A solvent-free protocol for one pot conversion of Baylis Hillman acetates to pyridopyrimidinones. Heterocyclic. Chemistry, v.48, n.3, p. 586-591, 2011.
STORK, G.; TAKAHASHI,T.; KAWAMOTO,I.; SUZUKI, T.; Total synthesis of prostaglandin F2α by Chirality Transfer from D-glucose. Journal of the American Chemical Society , v. 100, n. 26, p. 8272-8273, 1978.
TRAZZI, G.; ANDRÉ, M. F.; COELHO, F. Diastereoselective Synthesis of β-Piperonyl-γ-Butyrolactones from Morita-Baylis-Hillman Adducts. Highly Efficient Synthesis of (±)-Yatein, (±)-Podorhizol and (±)-epi-Podorhizol. Journal of the. Brazilian Chemical. Society, v. 21, n. 12, p. 2327-2339, 2010.
WASNAIRE, P.; WIAUX, M.; TOUILLAUX, R.; MARKÓ, I. E. Reductive cyclisation of Morita−Baylis−Hillman adducts. A simple approach towards substituted hydrindanones and decalones. Tetrahedron Letters, v. 47, p. 985–989, 2006.










