ABORDAGEM DA QUÍMICA MEDICINAL PARA A DESCOBERTA DE FÁRMACOS COM ATIVIDADE ANTI-INFLAMATÓRIA NO RECEPTOR DE ADENOSINA TIPO 2 (A2AR)
ISBN 978-85-85905-23-1
Área
Iniciação Científica
Autores
Silva, L.B. (UNIVERSIDADE FEDERAL DO AMAPÁ (UNIFAP)) ; Santos, K.L.B. (UNIVERSIDADE FEDERAL DO AMAPÁ (UNIFAP)) ; Ferreira, E.F.B. (UNIVERSIDADE FEDERAL DO AMAPÁ (UNIFAP)) ; Silva, C.H.T.P. (UNIVERSIDADE DE SÃO PAULO (USP)) ; Borges, R.S. (UNIVERSIDADE FEDERAL DO PARÁ (UFPA)) ; Santos, C.B.R.S. (UNIVERSIDADE FEDERAL DO AMAPÁ (UNIFAP))
Resumo
A inflamação é um mecanismo homeostático abstruso gerado para proteger a integridade do organismo contra agentes nocivos endógenos ou exógenos. O presente trabalho tem como objetivo a busca de novos candidatos a anti-inflamatório via triagem virtual baseado em farmacóforo, determinação das propriedades farmacocinéticas, toxicológicas e a predição sobre o sítio do metabolismo de uma molécula onde ocorre uma reação metabólica. O composto ZINC04257548 foi avaliado no citocromo P450 3A4 (CYP3A4), citocromo P450 2C9 (CYP2C9) e citocromo P450 2D6 (CYP2D6) isoenzima.
Palavras chaves
inflamação; farmacóforo; metabolismo
Introdução
A adenosina é um nucleósido endógena ubíqua que regula uma série de processos fisiológicos. Estes efeitos são mediados pela interação de ligação com receptores de adenosina (ARs) que são membros do receptor acoplado a proteína G (GPCR). Eles são conhecidos para serem quatro subtipos distintos AR, que são denominados A1, A2A, A2B e A3 (MEDZHITOV, 2008). O candidato a fármaco UK-432097 foi desenvolvido por Pfizer para o tratamento de doença pulmonar obstrutiva crônica, mas infelizmente deu resultados insuficientes de eficácia na fase II de ensaios clínicos (atividade cancerígena) (E; JACOBSON, 2011). As estruturas cristalinas de alta resolução resolvidas do complexo do sítio ativo hA2AR com os agonistas UK-432097 e 5'-N-etilcarboxamidoadenosina nos dão uma oportunidade única de colocar essas descobertas anteriores em um contexto atômico 3D e usar o conhecimento de interações atômicas para predizer novas ligandos substituídos (TOSH et al, 2012). Citocromos P450 (CYP), formam uma família de proteínas onipresente de enzimas heme-tiolato, que possuem 60 isoformas diferentes em humanos. Eles realizam uma série de reações diferentes, como alifática hidroxilação, N-, S- e O-desalquilação, oxidação aromática, epoxidação e oxidação de S e N (EKROOS, 2006). E, que também são a causa da maioria das interações medicamentosas e problemas de toxicidade dependentes do metabolismo, são divididos em reações de Fase I (oxidação, hidrólise, redução) e Fase II (conjugação). Assim, a previsão do (s) local (is) do metabolismo (SOMs) é um passo crucial no desenvolvimento de medicamentos (RYDBERG, 2010). Este trabalho se propõe a estudar os compostos com atividade agonista do receptor A2AR utilizando a metodologia de triagem virtual baseado em farmacóforo e predição metabólica no sítio metabólito citocromo P450.
Material e métodos
Os compostos foram selecionados a partir de uma pesquisa na base de dados BindingDB e Protein Data Bank (PDB), onde o composto UK-432097 e 20 agonistas A2AAR selecionados apresentam valores EC50 variando entre 0,66 e 2242,00 nM. A estrutura cristalográfica selecionada neste estudo extraída do PDB de código 3QAK complexada com o composto UK-432097. A seleção foi realizada para construir um modelo farmacofórico, com uso em experimentos de triagem virtual em bancos de dados de compostos comerciais. Todas as hipóteses farmacofóricas foram submetidas à validação externa de uma pequena subcoleção contendo 5 agonistas A2AAR e 15 antagonistas A2AAR (chamados "contaminantes"), bem como 500 compostos diversos. Assim, o modelo validado foi utilizado para triagem virtual baseado em farmacóforo nos bancos de dados ChemBrigde_DIVERSet, ChemBrigde_DIVERSet_Exp, zinc_Drug, Zinc_Natural_Stock e Zinc_FDA_BindingD no programa Discovery Studio, usando o melhor modelo farmacofórico obtido como restrição para pesquisar a base de dados. Os melhores compostos classificados em relação às restrições farmacofóricos foram filtradas em termo de predições das propriedades farmacocinética (ADME) usando o programa QikProp e toxicológica usando o programa DEREK, respectivamente. Posteriormente, o modelo farmacofórico validado e filtrado ADME/Tox foi submetido no web servidor SMARTCyp (http://www.farma.ku.dk/smartcyp/) ao qual usa um conjunto de energias de ativação pré-calculadas para fragmentos moleculares em combinação com descritores topológicos para a predição no sítio do metabolismo (SOM).
Resultado e discussão
O modelo farmacofórico escolhido usando o programa Discovery Studio foi baseado no ligante cristalográfico do PDB (agonista UK-432097- PDB 3QAK) e um grupo de 20 ligantes do BindingDB selecionados. O modelo farmacofórico mais eficaz selecionado contém três características (1 grupo hidrofóbico, 1 doador de ligação de hidrogênio e 1 aromático) colocadas no domínio de ligação A2AAR contendo 7 resíduos de aminoácidos (HIS264, THR256, ASN253, PHE168, SER277, HIS278 e THR88) cruciais para Atividade biológica. Através da triagem virtual resultou um total de 100 compostos, seguido por previsões ADME / Tox e inspeção visual, produzindo 6 novos agonistas A2AAR promissores para serem testadas in vitro ou in vivo, afim de confirmar a atividade anti-inflamatório, bem como as predições in silico. A predição metabólica foi realizada no composto ZINC04257548, ao qual o sítio do metabolismo (SOM) foi determinado nos átomos do composto que são modificados por enzimas (principalmente por P450) baseado na correspondência de fragmentos. Os resultados da isoenzima CYP3A4 / 2C9 / 2D6 estão resumidos na Figura 1, O eixo y (score) representa as pontuações relativas do SMARTCyp para cada modelo de isoforma aplicado a cada composto. As pontuações são relativas e específicas à combinação modelo / composto e não podem ser comparadas entre si. O eixo x (Ranking Átomo) representa os átomos de carbono 22 (1), 2 (2) e 28 (3). O CYP3A4 e CYP2C9 foi significativamente menor (P <0,05) do que o CYP2D6 em relação ao composto, verificou-se que a inibição da isoenzima CYP era CYP3A4< CYP2D6< CYP2C9. Assim, em nosso modelo CYP3A4, a soma dos descritores e a reatividade têm peso semelhante.
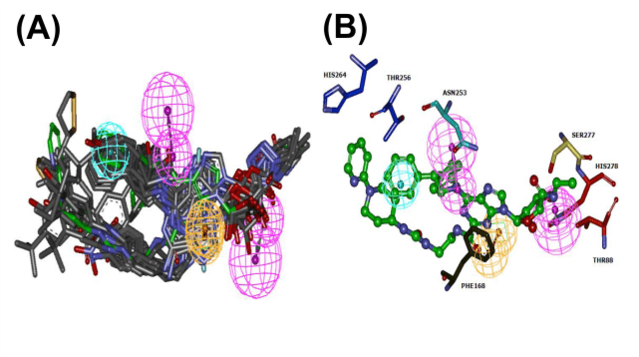
A) hipótese farmacofórica testados com um conjunto de compostos selecionados. B) hipótese farmacofórica e a análise do sitio ativo
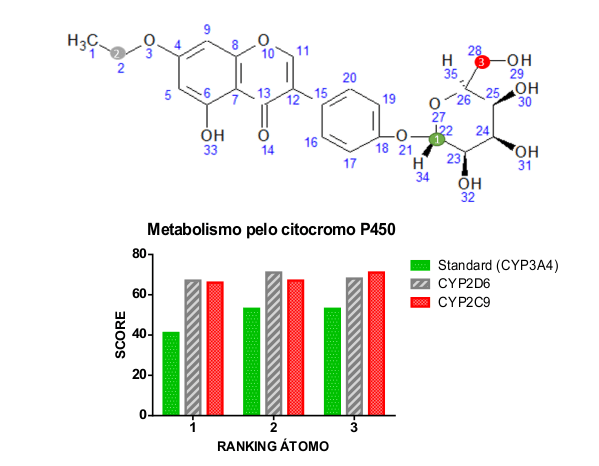
Análise do SMARTCyp da molécula ZINC04257548 utilizando os modelos padrão (CYP3A4), 2D6 e 2C9.
Conclusões
A predição no citocromo P450 são o CYP3A4, 2D6 e o 2C9, pelo composto ZINC04257548 oferece algumas vantagens clínicas, reduzindo o custo. O fato do SMARTCyp mostrar a reatividade é um fator principal no metabolismo do CYP 3A4. Os compostos selecionados apresentaram perfil farmacoterapêutico potencial e adequado, sugerindo seletividade e propriedades agonistas satisfatórias no A2AAR, o que pode estar correlacionado à atividade anti-inflamatório para serem testadas in vitro ou in vivo, afim de confirmar a atividade anti-inflamatório, bem como as predições in silico.
Agradecimentos
Ao programa CNPq/PIBIC pelo financiamento do projeto de pesquisa, à UNIFAP pela concessão da bolsa de Iniciação Científica e aos membros do Laboratório de Modelagem e Química Computacional (LMQC).
Referências
MEDZHITOV, R. Origin and physiological roles of inflammation. Nature, v. 454, p. 428 – 435, 2008.
HAUSLER, N. E.; DEVINE, S. M.; MCROBB, F. M.; WARFE L.; POUTON, C. W.; HAYNES, J. M.; BOTTLE, S. E.; WHITE, P. J.; SCAMMELLS, P. J. Synthesis and pharmacological evaluation of dual acting antioxidant A(2A) adenosine receptor agonists. Journal of Medicinal Chemistry, v.55, p. 3521-3534, 2012.
DI VIRGILIO, F. Purinergic mechanism in the immune system: A signal danger for dendritc cells. Purinergic Signalling, v. 1, p. 205 – 209, 2005.
E , C. E.; JACOBSON, K. A. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochimica et Biophysica Acta, v. 1808, p. 1290−1308, 2011.
TOSH, D. K.; PHAN, K.; GAO, Z .G.; GAKH, A. A.; XU, F.; DEFLORIAN, F.; ABAGYAN, R.; STEVENS, R. C.; JACOBSON, K. A.; KATRITCH, V. Optimization of Adenosine 5 Carboxamide Derivatives as Adenosine Receptor Agonists Using Structure-Based Ligand Design and Fragment Screening. Journal of Medicinal Chemistry, v. 55, p. 4297−4308, 2012.
Accelrys Discovery Studio 4.0, T.C., San Diego, CA 92121 USA.
LHASA LIMITED. Derek Nexus. Versão 10.0.2. Leeds, 2007
SCHRÖDINGER. QikProp: Rapid ADME predictions of drug candidates. Versão 3.4. New York, 2011.
EKROOS, M.; Sjogren, T. Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13682–13687.
RYDBERG, P. et al. SMARTCyp: a 2D method for prediction of cytochrome p450-mediated drug metabolism. Med. Chem. Lett. 1, 96–100, 2010.










