Propriedades eletrônicas do Nanotubo de GaN interagindo com as moléculas FeCl3 e CrO3 através de cálculos baseados na Teoria do Funcional da Densidade.
ISBN 978-85-85905-23-1
Área
Físico-Química
Autores
Silva, C.V.C.R. (UFMA) ; Coradi, S.G. (UFMA) ; Junior, J.J.G.V. (UFMA)
Resumo
Este trabalho apresenta um estudo teórico do nanotubo de GaN interagindo com as moléculas FeCl3 e CrO3 através de simulação de primeiros princı́pios, baseada na teoria do funcional da densidade, usando o software SIESTA. As propriedades estruturais, energéticas e eletrônicas das moléculas FeCl3 e CrO3 adsorvidas neste nanotubo foram analisadas, e constatou-se que as propriedades eletrônicas são afetadas pela interação das moléculas com o nanotubo de GaN. Além disso, novos nı́veis de energia apareceram na região do gap, diminuindo o gap de energia. Os cálculos também indicam que ambas as moléculas adsorvidas no exterior do nanotubo interagem com a superfı́cie através de um processo de adsorção quı́mica.
Palavras chaves
Nanotubo de GaN; DFT; Propriedades Eletrônicas
Introdução
No estudo de nanoestruturas, as simulações computacionais exercem um papel fundamental, visto que a síntese e a manipulação desses materiais exigem o uso de equipamentos caros, logo os estudos teóricos além de economizar reagentes, são capazes de predizer se a nanoestrutura existe ou não, e quais as propriedades físicas e químicas destes materiais. Nesse contexto, o estudo teórico de nanotubos tem sido um grande sucesso, pois vários nanotubos foram preditos antes da sua síntese (BLASE, 1994), como por exemplo, o nanotubo de Nitreto de Gálio (GaN). Este nanotubo pode ser descrito como uma folha de GaN do tipo grafeno (hexagonal) enrolada na forma cilíndrica com ao menos uma dimensão menor que 100nm. Os trabalhos, experimentais e teóricos, realizados com os nanotubos de nitreto de gálio, mostraram que este material é um semicondutor com largo gap de energia, em torno de 2,16 eV (LEE, 1999). Em razão disso, a aplicação deste material é limitada, e portanto, para potencializá-la é necessário a redução desse gap (CHERMAHINI et al., 2014). Com isso, o presente trabalho buscou reduzir o gap de energia do nanotubo de GaN interagindo as moléculas de FeCl3 e CrO3 na sua superfície externa através de simulações computacionais, a fim de aumentar a sua aplicabilidade.
Material e métodos
Para analisar as propriedades energéticas, estruturais e eletrônicas do nanotubo de GaN (10,0) interagindo com as moléculas de FeCl3 e CrO3, utilizou-se um nanotubo com 80 átomos (40 Ga e 40 N) mais os 4 átomos das moléculas, com célula unitária de 8,50 Å. Para tanto, realizou-se simulações computacionais de primeiros princípios fundamentadas na teoria do funcional da densidade (DFT) (HOHENBERG, 1964), as quais foram executadas no software computacional SIESTA (ARTACHO, 1999). As moléculas de FeCl3 e CrO3 foram colocadas em diferentes posições na superfície externa do nanotubo, o que resultou em 5 sistemas diferentes: 1. O átomo de Fe/Cr em cima do átomo de Ga; 2. O átomo de Cl/O em cima do átomo de N; 3. O átomo de Cl/O em cima do átomo de Ga; 4. O átomo de Fe/Cr no centro do hexágono; 5. O átomo de Cl/O no centro do hexágono. A otimização das geometrias, além dos cálculos de energia de ligação, distribuição de cargas e do gap de energia para o nanotubo de GaN interagindo com as moléculas também foram feitos no software SIESTA. Vale ressaltar que, o software estava configurado para utilizar a base dupla-zeta mais polarização (DZP) para representar a valência da função de onda. A interação dos elétrons do caroço e da valência foi descrita pelo pseudopotencial de Troullier-Martins (1991), e utilizou-se a aproximação do gradiente generalizado (GGA) para descrever o termo de troca e correlação (PERDEW et al., 1996).
Resultado e discussão
Os resultados obtidos foram satisfatórios, pois observou-se que em todas as
configurações o átomo
de Fe e Cr das moléculas migravam para cima do átomo de nitrogênio (N) do
nanotubo, formando uma
ligação intermolecular, com distâncias de 1,99 Å e 1,68 Å, respectivamente.
A energia de ligação
Fe-N e Cr-N calculada foi da ordem de -2,71 eV e -5,33 eV, respectivamente,
o que indica haver um
processo de adsorção química. As propriedades eletrônicas do nanotubo
interagindo com o FeCl3 e
CrO3 foram analisadas através da densidade de estados projetada (PDOS). Na
figura 1 (a) tem-se a
PDOS para o spin up e spin down do nanotubo de GaN puro, enquanto que na
figura 1 (b) a PDOS do
nanotubo de GaN com o FeCl3 adsorvido. O gap de energia foi calculado
através da PDOS, fazendo a
diferença de energia da banda de valência com a banda de condução. Assim,
para o nanotubo puro, o
gap de energia calculado foi de 2,35 eV, enquanto que para o nanotubo
interagindo com a molécula de
FeCl3 foi de 0,26 eV, pois o surgimento de novos níveis de energia provocou
a redução do gap. Além
disso, também houve o deslocamento do nível de fermi em 1,07 eV para a banda
de valência. De modo
análogo ao FeCl3, na PDOS do nanotubo de GaN interagindo com o CrO3 (Figura
2) também houve o
aparecimento de novos níveis de energia, o que reduziu o gap para 1,31 eV. E
além disso, o nível de
Fermi também foi deslocado em 0,82 eV para a banda de valência.
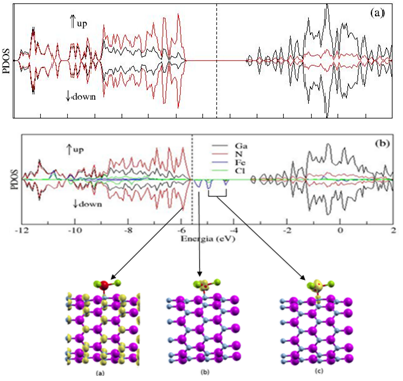
Densidade de estado projetada para o nanotubo de GaN (a) puro e (b) interagindo com a molécula FeCl3.
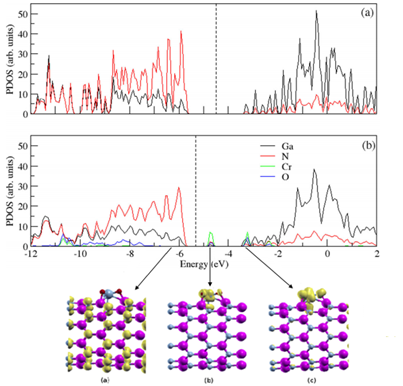
Densidade de estados projetada para o nanotubo de GaN (a) puro (b) intercalado com a molécula CrO3.
Conclusões
Os cálculos indicaram que ambas as moléculas se adsorvem na superfície do nanotubo por meio de um processo de quimissorção, onde há a formação de uma ligação entre o átomo de N do nanotubo e o átomo de Fe ou Cr das moléculas adsorvidas (Fe-N e Cr-N). E além disso, as propriedades eletrônicas do nanotubo são afetadas pela adsorção das moléculas, reduzindo o gap de energia para 0,26 eV, no caso da molécula de FeCl3, e para 1,31 eV, no caso da molécula de CrO3. Em razão disso, pode-se afirmar que novas possibilidades são abertas para aplicações deste material em nanodispositivos eletrônicos.
Agradecimentos
Referências
ARTACHO, E. et al. Linear-scaling ab-initio calculations for large and complex systems. Physica Status Solidi B-Basic Research, 215:809-817, 1999.
BLASE, X. at al. Stability and Band-gap Constancy of Boron Nitride nanotubes. Europhysics Letters, n. 2, p. 335-340, 1994.
CHERMAHINI, A. T. A. N.; FARROKHPOUR, H. Applied Surface Science, 320, 2014.
HOHENBERG, P.; KOHN, W. Inhomogeneous electron gas. Phys. Rev. B, USA, v. 136, p. B864, 1964.
LEE, S. M. et al. Stability and electronic of GaN nanotubes from density-functional calculations, Physical Review B, v. 60, n. 11, p. 7788-7791, 1999.
PERDEW et al. Generalized gradient approximation made simple. Physical Review Letters, 77(18):3865-3868, 1996.
TROULLIER N.; MARTINS, J. L. Physical Review B, 43:1993–2006, 1991.










