Modelo de solvatação de Grunwald-Winstein e solvólise do oxalato de bis(2,4,6-triclorofenila): mudanças no caráter do estado de transição com a composição de misturas etanol/água e metanol/água
ISBN 978-85-85905-25-5
Área
Química Orgânica
Autores
Soares, A. (UFABC) ; Bartoloni, F. (UFABC)
Resumo
Afim de melhor entender a influência do meio de solvatação na decomposição de ésteres oxálicos arílicos, comuns no eficiente sistema quimiluminescente peroxioxalato, utilizou-se o método de Grunwald-Winstein (GW) que avalia como o poder de ionização do solvente provoca mudanças nas cargas presentes no estado de transição. Empregando as constantes de velocidade de saída dos resíduos fenólicos (kobs1 e kobs2) os valores do fator de sensibilidade previstos por GW foram: 1,1 e 1,0 (100-95% EtOH/W), 0,196 e 0,3 (90-50% EtOH/W), 0,52 e 0,27 (100-70% MeOH/W), respectivamente, para kobs1 e kobs2. Foi postulado que em meios com maior quantidade de água o estado de transição (ET) é melhor estabilizado, consistente com um ET mais precoce.
Palavras chaves
Reação Peroxioxalato; Ésteres oxálicos; Grunwald-Winstein
Introdução
O conhecimento do mecanismo de uma reação é de extrema relevância, já que possibilita prever o resultado de uma transformação análoga, otimizando processos em busca de maiores rendimentos de preparação, possibilitar o entendimento de processos biológicos, entre outros. A descrição de um mecanismo de reação em termos de etapas elementares sempre é realizada de acordo com resultados cinéticos empíricos (CLAYDEN, GREEVES e WARREN, 2012). Nesse sentido, a físico-química orgânica utiliza ferramentas denominadas Correlações Lineares de Energia Livre (LFER, i.e., Linear Free Energy Relationships), que estão relacionadas à cinética de uma transformação, para a determinação de seu mecanismo (ANSLYN e DOUGHERTY, 2006). As LFER baseiam-se no princípio de que a energia de ativação de uma etapa ou a energia livre da reação mudam devido a uma alteração de substituinte no substrato ou solvente. Certos parâmetros quantitativos determinados experimentalmente correlacionam cada um desses efeitos químicos com a energia livre, tornando possível a avaliação de como tal alteração afeta o mecanismo de reação em termos de cinética. Por vezes, essas substituições podem levar a mudanças na superfície de energia da reação, favorecendo outro caminho reacional que não o estudado até o momento, levando a uma mudança de mecanismo (ANSLYN e DOUGHERTY, 2006). Uma LFER utilizada para avaliar a influência do meio de solvatação nas cargas presentes no estado de transição é a correlação de Grunwald-Winstein (GW), que permite a observação da participação ativa do solvente na reação (WINSTEIN, GRUNWALD e JONES, 1951). Deste modo, Grunwald e Winstein definiram uma nova escala baseada em uma reação de ionização, a solvólise de cloreto de t-butila (t-BuCl), que ocorre via mecanismo SN1, utilizando como meio solvolítico padrão uma mistura contendo 80% de etanol (EtOH) e 20% de água (W), em volume (80% EtOH/W). A transformação envolve a formação de um carbocátion terciário como intermediário na etapa determinante de velocidade (e.d.v.), seguida da adição de água ou etanol. A partir do logaritmo da razão entre as constantes de velocidade em um solvente qualquer e o meio padrão, determina-se a escala Y que é o poder de ionização do solvente (GRUNWALD e WINSTEIN, 1948). A escala de GW permite que esta metodologia seja aplicada para outras reações. A partir do logaritmo da relação entre as constantes de velocidade da reação estudada e constante de velocidade da mesma reação no meio solvolítico padrão é possível determinar um fator de sensibilidade m (Equação 1). A magnitude do valor de m fornece informações acerca da geração de carga no estado de transição da reação, bem como do grau de assistência nucleofílica do solvente (FAINGBERG e WINSTEIN, 1955). O método desenvolvido por GW é atualmente uma ferramenta bastante utilizada para auxiliar na racionalização de mecanismos (CHOI et al., 2017; CHOI e KOO, 2017; KOH e KANG, 2015). O método de GW foi utilizado nesse trabalho como ferramenta para melhor compreender a influência do meio de solvatação na decomposição do éster oxálico oxalato de bis(2,4,6-triclorofenila) (TCPO), para obter informações acerca da geração de carga no estado de transição da reação, e a importância do poder de ionização do solvente para a reação estudada. Tal éster é comumente empregado na reação peroxioxalato, que é um dos principais sistemas quimiluminescentes orgânicos com alto rendimento de emissão, levando a uma ampla aplicação analítica para detecção de baixas concentrações de analitos (STEVANI e BAADER, 2002). Tal transformação consiste em uma reação, catalisada ou não por base, entre um éster oxálico arílico (e.g., TCPO) e peróxido de hidrogênio na presença de um ativador (ACT). O mecanismo geral mais aceito para tal transformação envolve a formação de um intermediário de alta energia (IAE), seguida do consumo desse IAE pelo ACT, levando a formação de um estado excitado. O retorno do ativador excitado para o estado fundamental ocorre de forma radiativa, levando à emissão de luz que é um dos produtos da reação (CABELLO, EL SEOUD e BAADER, 2018; CISCATO et al., 2012). Em termos de eficiência de formação de estados excitados, é relevante determinar com qual eficiência um certo éster oxálico é convertido ao IAE, considerando-se efeitos de solvatação na reação do éster com nucleófilos e/ou na formação de perácidos intermediários. Usualmente, tais estudos são realisados por meio de medidas cinéticas de quimiluminescência, correlacionando-se constantes de velocidade com a concentração empregada de reagentes (REIS, 2018). Logo, o método de estudo baseado em GW é diferenciado para racionalização de tal transformação. Para isso, foram realizados ensaios cinéticos de absorção de luz para a solvólise do TCPO, em misturas binárias de metanol e etanol com água, possibilitando uma análise quantitativa da influência do meio de solvatação na decomposição do TCPO através do fator de sensibilidade m previsto no modelo de GW.
Material e métodos
Oxalato de bis(2,4,6-triclorofenila) (TCPO, Sigma-Aldrich, 99%) foi purificado por recristalização (Hexano:Acetato de Etila). Os solventes utilizados para os ensaios cinéticos foram etanol (EtOH, Sigma-Aldrich, ≥ 99,5%); metanol (MeOH, Sigma-Aldrich, ≥ 99,9%); água deionizada (W) obtida pelo sistema de purificação Milli-Q Millipore (conductividade igual à 18,2 MΩ) e para o preparo da solução estoque foi utilizado o acetato de etila (Sigma-Aldrich, ≥ 99%). Espectros de absorção na região do ultravioleta-visível (UV-Vis) e medidas cinéticas de absorção de luz foram obtidos no espectrofotômetro Agilent Cary 60 UV-Vis (Santa Clara, CA, EUA), equipado com multicell holder termostatizado por um banho Varian Cary PCB 1500l acoplado a um computador para armazenamento de dados. Foram utilizadas cubetas de quartzo para absorção com volume máximo de 3,0 mL e caminho óptico de 1,0 cm. Ensaios cinéticos foram realizados acompanhando a absorção de luz, decorrente da solvólise do éster (concentação do TCPO ulilizada como referência foi 0,1 mmol L–1 a 25 °C) em EtOH e MeOH, além de sua decomposição em misturas binárias de cada um dos solventes orgânicos com água. Assim as misturas estudadas foram: 100; 98; 95; 90; 80; 70; 60; 50% EtOH/W (v/v) e 100; 90; 80; 70% MeOH/W (v/v), sendo o fator limitante para a adição de maiores volumes de água a solubilidade do éster. O meio de reação de referência é a mistura de 80% EtOH/W, adotado como meio solvolítico padrão para o sistema de GW. Os espectros e perfis cinéticos de absorção de luz foram ajustados matematicamente empregando-se o software de tratamento de dados OriginPro 8.5 (OriginLab). A partir do perfil cinético de absorção, foi possível obter as constantes de velocidade para os processos de formação do fenol e/ou fenolato. Em todos os casos foram observados perfis cinéticos de saturação compostos por duas contantes de velocidade (kobs1 e kobs2), referentes à saída de dois resíduos fenólicos. De posse das constantes de kobs1 e kobs2, nos diferentes meios de solvatação, o fator de sensibilidade (m) previsto no modelo de GW, foi determinado por gráficos correlacionando as constantes de velocidade observada com os valores de Y, estes definidos para a transformação padrão (solvólise de t-BuCl em 80% EtOH/W). A influência da temperatura nas contantes de velocidade também foi avaliada, realizando a aquisição dos perfis cinéticos nas temperaturas de 21, 25, 30, 35 e 40 ºC. Deste modo, ao utilizar a equação de Arrhenius, foi possível obter os parâmetros de ativação da transformação e, a partir destes, os valores de entalpia (∆H‡), entropia (∆S‡) e de Energia Livre de Gibbis (∆G‡) de ativação, da decomposição de TCPO em todos os meios estudados.
Resultado e discussão
A decomposição do TCPO foi realizada em doze meios diferentes (100; 98; 95;
90; 80;70; 60; 50% EtOH/W e 100; 90; 80; 70 % MeOH/W), a qual foi
acompanhada no UV-Vis, para obtenção do perfil cinético de solvólise e das
constantes de velocidade kobs1 e kobs2. Ambas as constantes de velocidade
foram correlacionadas com o poder de ionização da mistura de solvente (Υ) e,
a partir de uma relação linear (r > 0,9) de GW, para etanol na composição de
100 a 95% EtOH/W do coeficiente angular foi obtido o fator de sensibilidade
(m) de 1,1 ± 0,3 e 1,0 ± 0,1, respectivamente, para kobs1 e kobs2.
Entretanto, na composição de 90 a 50% EtOH/W obteve-se m = 0,196 ± 0,009 e
0,30 ± 0,01, também para kobs1 e kobs2. Em metanol, foi observado um único
comportamento linear, com um valores de m = 0,52 ± 0,05 e 0,27 ± 0,01,
respectivamente, para kobs1 e kobs2.
Tais valores de m indicam que de 100-95% EtOH/W a reação é extremamente
sensível a habilidade de ionização do meio tendo uma total ionização na
e.d.v., já entre 90-50 % EtOH/W a reação se torna menos sensível às mudanças
na composição do meio. O valor de m obtido para as misturas de MeOH/W,
considerando primeiramente kobs1, foi um valor intermediário aos obtidos
para as duas regiões de EtOH, já para kobs2 observou-se um coeficiente
análogo ao 90-50% EtOH/W. Apesar disso, pode-se concluir que a saída dos
dois resíduos fenólicos em metanol demonstraram baixa sensibilidade ao
efeito do solvente.
De posse dos valores de m foram propostos dois mecanismos distintos, o
primeiro considerando uma total ionização na e.d.v, com um estado de
transição (ET) mais tardio no qual há uma maior formação de carga positiva
no oxigênio proveniente do nucleófilo e carga formal negativa no oxigênio
originário da carbonila, sendo que tal ET ocorre em misturas 100-95% EtOH/W
(Figura 1). Já a segunda proposta dispõe de menor geração de carga na e.d.v.
e consequente ET mais precoce quando comparado com a primeira proposição,
sendo válida em misturas 90-50% EtOH/W e MeOH/W (Figura 1).
A fim de obter maiores evidências acerca do mecanismo de tal transformação,
foram calculados os parâmetros de ativação da reação em todos os meios,
sendo que em todos os casos a Ea e o ∆H‡ não seguiram uma tendência linear,
sendo mais relevantes as comparações em termos de ∆G‡ e ∆S‡. Deste modo, os
valores de ∆G‡ em EtOH/W variaram de 21,6 ± 0,1 a 19,4 ± 0,4 kcal mol–1 e de
23,9 ± 0,5 a 21,7 ± 0,2 kcal mol–1 (para kobs1 e k obs2 respectivamente) e a
variação de ∆S‡ foi de –50,2 ± 0,2 a –44,4 ± 0,4 cal mol–1 K–1 e de –47 ± 1
a –43 ± 0,3 cal mol–1 K–1 (para kobs1 e kobs2 respectivamente). Em MeOH/W os
valores de ∆G‡ variaram de 20,3 ± 0,8 a 18,8 ± 0,1 kcal mol–1 e de 21,5 ±
0,4 a 20,9 ± 0,4 kcal mol–1 (para kobs1 e k obs2 respectivamente), e a
variação do ∆S‡ foi de –44 ± 2 a –39,0 ± 0,4 cal mol–1 K–1 e de –41,2 ± 0,9
a –43,9 ± 0,9 cal mol–1 K–1 (para kobs1 e kobs2 respectivamente). Os valores
obtidos corroboram as propostas mecanísticas para o caráter do ET, uma vez
que em EtOH/W foram observados valores extremamente elevados de ∆G‡
(principalmente de 100-95% EtOH/W), que corroboram um ET tardio, e um ∆S‡
extremamente negativo indicando um processo associativo.
Para melhor compreender o motivo pelo qual na região entre 100-95% EtOH/W a
solvólise de TCPO é extremamente sensível ao efeito do meio, deve-se
considerar que em misturas binárias de solventes há uma micro-
heterogeneidade, uma vez que ao adicionar água em um solvente polar prótico
(ROH; EtOH ou MeOH) puro altera-se a organização das moléculas do solvente
orgânico até um ponto que a as interações entre ROH-W são extremamente
importantes para a composição geral do meio, sendo que com o aumento da
poncentagem de água os complexos envolvendo apenas moléculas de água tornam-
se cada vez mais relevantes. O conhecimento da identidade do meio é de
extremo valor, já que altera a polaridade do meio, bem como a esfera de
solvatação do éster e do estado de transição da transformação em questão (EL
SEOUD, 2009). Bastos et al., utilizando uma sonda solvatocrômica, puderam
averiguar que a polaridade de EtOH é consideravelmente menor do que de MeOH,
e a diferença da polaridade entre o EtOH e W é significantemente maior do
que entre MeOH e W (BASTOS, SILVA e EL SEOUD, 2006).
No caso da solvólise de TCPO, pode-se supor que MeOH têm o poder de solvatar
o estado de transição de maneira mais efetiva que o etanol. Em vista disso,
ao adicionar uma quantidade significativa de água ao MeOH não há mudanças
consideráveis na energia do ET, no entanto, em EtOH a mudança na composição
do meio é muito mais significativa, exatamente pelo aparecimento de
complexos de água que estabilizam de maneira mais efetiva o ET (BASTOS,
SILVA e EL SEOUD, 2006). Adicionalmente, como a quantidade de complexos
MeOH-W é maior do que complexos análogos EtOH-W, como visto também por
Bastos et al., (BASTOS, SILVA e EL SEOUD, 2006) a quantidade de complexos
envolvendo apenas moléculas de água ocorre em maior proporção em EtOH,
provocando alterações mais significativas sentidas pelo ET da solvólise. Tal
mudança pode ser visualizada tanto na correlação de GW quanto nas alterações
do comportamento linear do ∆G‡, uma vez que com a diminuição significativa
da barreira de ativação o ET torna-se mais precoce.
Outro resultado interessante é a variação considerável do valor de m em
metanol, de 0,52 ± 0,05 a 0,27 ± 0,01 (de kobs1 para kobs2), que pode ser
atribuído à menor reatividade da saída do segundo resíduo fenólico, a qual é
observada principalmente no aumento do valor de ∆G‡ em comparação ao de
kobs1. Tal diminuição de velocidade provavelmente é proveniente da perda do
grupo arílico que, por efeito indutivo sacador de elétron, torna o carbono
carbonílico mais eletrodeficiente para a ocorrência da etapa de adição.
Deste modo, após a eliminação do grupo de partida, a reação se torna mais
lenta para a saída do segundo resíduo fenólico e menos sensível à natureza
do meio (NEUVONEN, 1994).
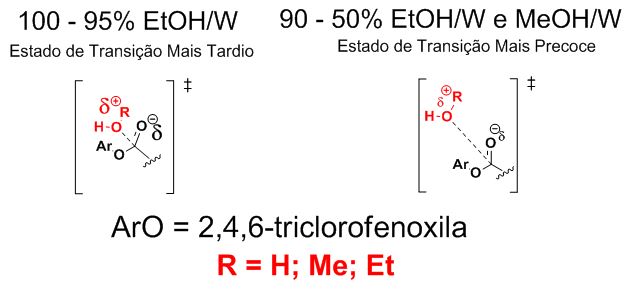
Estado de transição mais precoce para a transformação em 100-95% EtOH/W e mais tardio em 90-50% EtOH/W e MeOH/W.
Conclusões
A partir dos valores de fator de sensibilidade m previsto por GW foi possível averiguar a natureza do ET da solvólise de TCPO. O valor de m = 1,1 ± 0,3 e 1,0 ± 0,1 em 100-95% EtOH/W, para kobs1 e kobs2, respectivamente, permite concluir que nesse intervalo de composição a reação é extremamente sensível a habilidade de ionização do meio, tendo uma total ionização na e.d.v e um estado de transição mais tardio. Entretanto, na composição de 90- 50% EtOH/W os valores de m obtidos foram 0,196 ± 0,009 e 0,30 ± 0,01 para kobs1 e kobs2, respectivamente, mostrando que a reação se torna menos sensível as mudanças na composição do meio e com um estado de transição mais precoce. Os valores obtidos de ∆G‡ em EtOH/W corroboram as propostas de natureza do ET. Em MeOH, os valores de m obtidos são 0,52 ± 0,05 e 0,27 ± 0,01, sendo que a reação é pouco sensível às mudanças na composição do meio e o ET é mais precoce com valores de ∆G‡ e ∆S≠ condizentes para tal. A mudança observada na correlação de GW para EtOH/W ocorre devido a diminuição significativa da energia do ET decorrente da maior contribuição da água para sua solvatação. Em metanol tal desvio não é observado, já que complexos de água estão presentes em menor quantidade em MeOH/W, além de MeOH solvatar melhor o ET do que EtOH, fazendo com que a mudança na composição do meio seja muito menos percebida pelo ET da reação. Em vista dos resultados obtidos é relevante realizar o mesmo tipo de estudo utilizando outros solventes polares próticos para enfim racionalizar a aplicação do TCPO nesses meios, além de verificar o comportamento com a adição de nucleófilos diversos ‒ e.g., imidazol, H2O2 e azida. Tais informações são particularmente relevantes no contexto da reação quimiluminescente peroxioxalato, devido seu reconhecido valor analítico.
Agradecimentos
O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - Código de Financiamento 001.
Referências
ANSLYN, E. V.; DOUGHERTY, D. A. Modern physical organic chemistry. University science books, 2006.
BASTOS, E. L.; SILVA, P. L.; EL SEOUD, O. A. Thermosolvatochromism of Betaine Dyes Revisited: Theoretical Calculations of the Concentrations of Alcohol− Water Hydrogen-bonded Species and Application to Solvation in Aqueous Alcohols. The Journal of Physical Chemistry A, v. 110, n. 34, p. 10287-10295, 2006.
CABELLO, M. C.; EL SEOUD, O. AA; BAADER, W. J. Effect of ionic liquids on the kinetics and quantum efficiency of peroxyoxalate chemiluminescence in aqueous media. Journal of Photochemistry and Photobiology A: Chemistry, v. 367, p. 471-478, 2018.
CHOI, H.; KOO, In S. Mechanistic Study of the Solvolysis of Piperidine‐1‐sulfonyl chloride in Binary Solvent Mixtures. Bulletin of the Korean Chemical Society, v. 38, n. 9, p. 1105-1108, 2017.
CHOI, H. et al. Kinetic Study on Solvolysis of 2‐Methylfuran‐3‐carbonyl Chloride in Binary Solvent Mixtures. Bulletin of the Korean Chemical Society, v. 38, n. 3, p. 320-323, 2017.
CLAYDEN, J..; GREEVES, N.; WARREN, S. Organic Chemistry, 2nd ed., Oxford University Press, 2012.
CISCATO, L. F. ML et al. The chemiluminescent peroxyoxalate system: state of the art almost 50 years from its discovery. Arkivoc, p. 391-430, 2012.
EL SEOUD, O. A. Understanding solvation. Pure and Applied Chemistry, v. 81, n. 4, p. 697-707, 2009.
FAINBERG, A. H.; WINSTEIN, S. Salt Effects and Ion Pairs in Solvolysis and Related Reactions. V. Special Salt Effect in Acetolysis of 2-Anisylethyl p-Toluenesulfonates. Journal of the American Chemical Society, v. 78, n. 12, p. 2767-2770, 1956.
GRUNWALD, E.; WINSTEIN, S. The correlation of solvolysis rates. Journal of the American Chemical Society, v. 70, n. 2, p. 846-854, 1948.
KOH, H. J.; K., S. J. Correlation of Rates of Solvolysis of Diphenylacetyl Chloride Using Extended Grunwald–Winstein Equation. Bulletin of the Korean Chemical Society, v. 36, n. 10, p. 2429-2433, 2015.
REIS, R.. A. Fluorescência e Quimiluminescência para a Determinação de Mecanismos de Reação na Decomposição de Ésteres. 2018. 109 f. Dissertação (Mestrado em Química) - Universidade Federal do ABC, São Paulo, 2018.
STEVANI, C. V.; BAADER, W. J. Preparation and Characterization of 2,2,2-triphenyl-2λ 5-1,3,2-dioxastilbolane-4,5-dione as Standard for an Attempt to Trap 1,2-Dioxetanedione, a Possible High-Energy Intermediate in Peroxyoxalate Chemiluminescence. Journal of Chemical Research, p. 430-432, 2002.
NEUVONEN, H. Kinetics of the decomposition of a chemiluminescent reagent bis (2, 4-dinitrophenyl) oxalate in aqueous acetonitrile. Journal of the Chemical Society, Perkin Transactions 2, n. 1, p. 89-95, 1994.
WINSTEIN, S.; GRUNWALD, E.; JONES, H. W. The correlation of solvolysis rates and the classification of solvolysis reactions into mechanistic categories. Journal of the American Chemical Society, v. 73, n. 6, p. 2700-2707, 1951.








