
Estudo Computacional dos Orbitais Naturais de Ligação e Avaliação Farmacocinética e Toxicológica in silico para o Composto Dietilditiocarbamato de Fe(II)
- Home
- Trabalhos
ÁREA
Físico-Química
Autores
Téllez Zepeda, C.A. (UNIVERSIDADE BRASIL) ; Pessoa, R.S. (UNIVERSIDADE BRASIL) ; Téllez Soto, C.A. (UNIVERSIDADE BRASIL) ; Costa Jr., A.C. (INSTITUTO FEDERAL DO RIO DE JANEIRO IFRJ) ; Versiane, O. (INSTITUTO FEDERAL DO RIO DE JANEIRO IFRJ)
RESUMO
O modelo dos orbitais naturais descreve o conjunto exclusivo de funções ortonormais de um elétron intrínsecas à função de onda de n elétrons e expressa como os orbitais atômicos formam as ligações moleculares características. Neste trabalho, empregamos a análise dos orbitais naturais de ligação (NBOs) com o nível de teoria do funcional da densidade (DFT) para obter informações sobre a geometria molecular e sobre as funções de onda das ligações químicas presentes no composto de coordenação Dietilditiocarbamato de Fe(II). Os dados calculados para a geometria do composto estão de acordo com os dados experimentais obtidos pelos autores que sintetizaram o composto por primeira vez em 1975. Realizamos, ainda, predições farmacocinéticas e toxicológicas in silico preliminares para o composto.
Palavras Chaves
NBO; DFT; Ditiocarbamatos
Introdução
Ditiocarbamatos (DTCs) são compostos de coordenação organossulfurados que formam complexos estáveis com metais mediante a estabilização de cátions metálicos (ODULARU & AJUBADE, 2019 ; MENEZES & LIMA, 2021). Os DTCs podem apresentar diversas aplicações, dentre as quais a literatura destaca seu emprego como pesticidas, fungicidas, agentes antitumorais e biocidas (MENEZES & LIMA, 2021), além de outras aplicações biomédicas, tais como ação antitumoral (MANAV et al, 2006), imunomoduladora (TOPPING & JONES, 1988), antidiabética (YOSHIKAWA et al, 2007) e antiviral (UCHIDE & OHYAMA, 2003). O complexo [Fe(DDTC)2] foi sintetizado originalmente por Ileperuma e Feltham, que determinaram sua estrutura por difração de raios X (ILEPERUMA & FELTHAM, 1975). No que diz respeito a aplicações biomédicas, complexos de Fe(II) coordenados com dietilditiocarbamato têm sido utilizados para a detecção de óxido nítrico (NO) em membranas celulares (VANIN et al, 1993) e em vasos sanguíneos isolados (KLESCHYOV & MÜNZEL, 2002), o que é pertinente para o possível desenvolvimento de abordagens farmacológicas para condições tais como arteriosclerose, hipertensão e diabetes. Ademais, complexos de ditiocarbamatos com metais de transição têm sido reportados na literatura como possíveis inibidores de proteassoma (CVEK & DVORAK, 2007), o que os torna potenciais candidatos para o desenvolvimento de terapias antitumorais. Dessa maneira, nosso estudo teórico da estrutura eletrônica do complexo [Fe(DDTC)2] é pertinente, uma vez que a compreensão de suas características estruturais é necessária para orientar o potencial desenvolvimento futuro de aplicações biomédicas ou industriais ainda não conhecidas para o composto específico deste estudo. Realizamos um trabalho exclusivamente teórico, portanto não produzimos dados experimentais. Nosso objetivo consiste em aplicar a análise computacional dos Orbitais Naturais de Ligação (NBO) utilizando a Teoria do Funcional de Densidade (DFT) para elucidar características da estrutura eletrônica do composto de coordenação [Fe(DDTC)2], bem como a constituição dos orbitais nas ligações centrais que o composto apresenta (Fe-S, N=C, C-S e C- C). Como objetivo secundário e meramente ilustrativo, realizamos algumas predições farmacocinéticas e toxicológicas preliminares para o [Fe(DDTC) 2], empregando métodos in silico. Justifica-se a escolha do nível de teoria do DFT por apresentar concordâncias melhores com resultados experimentais em comparação com os métodos Hartree-Fock e pós-Hartree-Fock, além de ganhos em custo computacional (KOCH & HOLTHAUSEN, 2001). Já o conceito de orbitais naturais foi introduzido em 1955 e descreve o conjunto exclusivo de funções ortonormais de um elétron que são intrínsecas à função de onda de n elétrons (LÖWDIN, 1955). A análise dos NBO, fundamentada nos orbitais naturais, relaciona soluções computacionais para a função de onda de Schrödinger com conceitos acerca de ligações químicas (WEINHOLD et al, 2016). Dessa maneira, escolhemos esse método uma vez que proporciona informações a respeito da localização dos orbitais atômicos naturais em uma molécula, do grau de deslocalização de seus elétrons e do balanço de cargas, bem como de outros aspectos relativos à sua estrutura eletrônica. Dessa maneira, o método de análise dos NBO possibilita obter uma transformação otimizada de uma função de onda em uma forma localizada, correspondente a elementos de um centro, correspondente a um par isolado, e de dois centros, correspondentes a uma ligação de acordo com a estrutura de Lewis, o que proporciona informações relevantes a respeito da natureza das ligações químicas entre metais de transição e ligantes em compostos de coordenação (VEKTARIENE, 2018). Enfatizamos que nosso método de análise dos NBO utilizando a DFT com o funcional híbrido B3LYP e conjunto de bases 6-311G(d,p) foi validado em diversos artigos de pesquisa já publicados por alguns autores deste trabalho para a comparação entre dados experimentais e teóricos referentes tanto a geometrias moleculares obtidas por difração de raios X para monocristais quanto aos números de onda vibracionais em compostos de coordenação estruturalmente semelhantes ao [Fe(DDTC)2]: [Mn(DDTC) 2] (TÉLLEZ et al, 2016); [Cd(DDTC)2] (TÉLLEZ et al, 2015); [Zn(DDTC)2] (COSTA et al, 2013); [Cu(DDTC)2] (COSTA et al, 2013b); e [Co(DDTC)2] (NEVES et al, 2012). Isso reforça nossa escolha do nível de teoria e do método para o presente estudo. Para as predições farmacocinéticas e toxicológicas do composto [Fe(DDTC) 2], obtivemos, in silico, alguns de seus dados farmacocinéticos ADME (absorção, distribuição, metabolismo e excreção) e características toxicológicas. A realização de estudos in silico preliminares reduz a possibilidade de falhas nos estágios subsequentes do desenvolvimento de um potencial fármaco, permitindo assim redução de custos.
Material e métodos
Para nossa análise teórica empregando o método NBO para o composto [Fe(DDTC) 2], utilizamos o nível de teoria do funcional de densidade (DFT) para química quântica computacional, com a função de base correspondente ao método do funcional híbrido B3LYP, introduzido por Axel Becke nos parâmetros Lee-Yang-Parr (MARSMAN et al, 2008 ; LEE, 1988), com o conjunto de bases 6- 311G(d, p). Realizamos nossos cálculos utilizando modelagem computacional com o software Gaussian 09W (GAUSSIAN, 2013) em um notebook Acer Aspire E5-573, Intel@ Core(TM) i5-5200U (CPU de 2.20 GHz, dual-core), com 16 GB de memória RAM e sistema operacional Microsoft Windows 10 Pro. Para realizar as predições farmacocinéticas e toxicológicas do composto [Fe(DDTC)2], meramente preliminares e ilustrativas, utilizamos o servidor online PreADMET (PREADMET, 2017).
Resultado e discussão
Obtivemos a geometria para o [Fe(DDTC)2] (Figura 1) utilizando o
software Chemcraft (ANDRIENKO, 2019) e realizamos a otimização mediante o
procedimento B3LYP/6-311G(d,p). As distâncias interatômicas e os ângulos
calculados (calc.) por DFT concordam com os valores experimentais (exp.)
obtidos pelos autores que sintetizaram o composto (ILEPERUMA & FELTHAM,
1975).
As distâncias para as ligações entre o átomo de Fe central e os átomos de S
circundantes foram de 2,299 Å (calc.) e 2,452 Å (exp.) para Fe28-S25, 2,270
Å (calc.) e 2,402 Å (exp.) para Fe28-S26, 2,269 Å (calc.) e 2,408 Å (exp.)
para Fe28-S29 e 2,297 Å (calc.) e 2,437 Å (exp.) para Fe28-S27. Para as
ligações entre os átomos de N e C, obtivemos as distâncias 1,337 Å (calc.) e
1,321 Å (exp.) para N32-C24 e 1,335 Å (calc.) e 1,330 Å (exp.) para N31-C30.
Para as ligações entre os átomos de C e S, obtivemos as distâncias 1,725 Å
(calc.) e 1,766 Å (exp.) para C24-S27, 1,751 Å (calc.) e 1,721 Å (exp.) para
C24-S29, 1,751 Å (calc.) e 1,731 Å (exp.) para C30-S26 e 1,725 Å (calc.) e
1,734 Å (exp.) para C30-S25. Obtivemos, ainda, os ângulos de
101,420o (calc.) e 102,3o (exp.) para S29-Fe28-S25,
50,239o (calc.) e 74,3o (exp.) para S25-Fe28-S26,
101,368o (calc.) e 98,3o (exp.) para S27-Fe28-S26,
112,589o (calc.) e 114,7o (exp.) para S27-C24-S29,
112,742o (calc.) e 115,7o (exp.) para S26-C30-S25.
Observamos que os dados experimentais foram obtidos por Ileperuma e Feltham
(1975) a partir da estrutura cristalina, enquanto nossos cálculos foram
feitos sobre a molécula isolada.
Na análise dos NBO para as estruturas naturais de Lewis, dados dois átomos A
e B, cada tipo de NBO A-B se decompõe em seus orbitais híbridos
constituintes naturais (NHOs), hA e hB, de acordo com ΨAB = cA⋅hA + cB⋅hB. Os
coeficientes de polarização cA e cB satisfazem a equação |cA|
2+|cB|2 = 1. Os híbridos de ligação hA e hB se
assemelham à descrição clássica de hibridização de Pauling, porém tanto os
detalhes da hibridização pelos NHOs quanto as polarizações são otimizadas
numericamente pelo método para proporcionar a melhor descrição possível da
densidade eletrônica. Os NBOs de duas camadas de valência são definidos
pelos híbridos de valência hA e hB, uma em fase do tipo Lewis, dada por ΨAB
= cA⋅hA + cB⋅hB, e o NBO fora de fase (não ocupado) correspondente, que não é
do tipo Lewis e é dado por Ψ*AB = cB⋅hA-cA⋅hB.
Quanto aos orbitais e às matrizes de densidade, o conjunto de NBOs do tipo
Lewis costuma incluir um núcleo central CR e um par de valência solitária
LP, assim como orbitais de ligação de dois centros, BD. O conjunto de Lewis
inclui ainda a valência não ocupada sem ligação, LP* e a camada
de valência extra dos orbitais de Rydberg, RY*, assim como os
antiligantes de valência BD* que vêm a partir da equação Ψ*
AB = cB⋅hA-cA⋅hB. Dessa maneira, os NBOs constituem um conjunto de
bases químicas do tipo Lewis e também orbitais que não correspondem ao tipo
Lewis, sendo que cada membro se encontra relacionado ao diagrama da
estrutura localizada de Lewis ou à sua capacidade de alteração química
(WEINHOLD & LANDIS, 2005 ; WEINHOLD et al, 2016 ; GLENDENING et al, 2013 ;
LANDIS & WEINHOLD, 2016).
Considerando a interação dos elétrons, as ligações Fe-S são formadas pelas
seguintes interações: φS26F28 = 0,9158(sp11,82d0,02)
S26 + 0,4017(sp0,03d1,12)Fe28
com polarização de 83,87% para o S26 e 16,13% para o Fe28, e ocupação
eletrônica de 1,94180 elétrons, caracterizando uma ligação simples; e
φS27F28 = 0,9179(sp12,74d0,02)S27 +
0,3963(sp0,03d1,09)Fe28, com polarização de
84,25% para S27 e 15,76% para Fe28 e ocupação eletrônica de 1,93724.
A análise mostra que a ligação N32-C24 apresenta características de ligação
simples. No entanto, a ligação N31—C30 se mostra como ligação dupla. No
espectro vibracional, o modo vibracional de estiramento N32-C24 apresenta
deslocamento de 10 cm-1, com número de onda menor do que a
vibração de estiramento da ligação N31-C30. Com relação ao deslocamento
eletrônico entre os átomos que formam os dois anéis quadrados planos com o
átomo de Fe no centro, a ligação C24-S27 se mostra como ligação dupla.
Entretanto, as demais ligações C-S se apresentam como ligações simples. No
espectro vibracional, o modo de estiramento C24-S27 se localiza em 610
cm-1 (calc.). Já para os estiramentos C24-S29, C30-S26 e C30-S25,
os valores de números de onda são 567 e 579 cm-1 respectivamente,
sendo o segundo valor correspondente a uma vibração simétrica. A
distribuição de valência dos átomos de C24 e C30 é concordante.
A ligação C30-N31, que se apresenta como ligação dupla, apresenta ocupação
eletrônica total de 3,95 elétrons e as funções de onda para essa ligação
são: φC30N31 = 0,6031(sp1,83d0,00)C30 +
0,7961(sp1,62d0,00)N31, com polarização de
36,62% sobre o átomo de C30 e de 63,38% sobre o átomo N31, e φC30N31 =
0,5186(sp1,00d0,00)C30 +
0,8550(sp1,00d0,00)N31, com polarização de
26,89% para o átomo de C30 e de 73,11% para N31, valores concordantes com a
eletronegatividade dos átomos.
Quanto às transferências de cargas, as interações doadoras e aceptoras
decorrem da análise da matriz de Fox, segundo a teoria da perturbação de
segunda ordem. As interações mais representativas para o complexo [Fe(DDTC)
2], obtidas por intermédio dos cálculos de NBO/B3LYP, são as
seguintes: entre o orbital doador (OD) 95 (LP (3) S29) e o orbital aceptor
(OA) 477 (BD*( 1) C24-N32), com energia de 10,18 Kcal/mol; entre
o OD 94 (LP (2) S29) e o OA 479 (BD*(1) S26-Fe28), com 11,06
Kcal/mol; entre o OD 81 (LP (2) S25) e o OA 482 (BD*(1) C30-N31),
com 12,23 Kcal/mol; entre o OD 94 (LP (2) S29) e o OA 475 (BD*(2)
C24-S27), com 46,35 Kcal/mol; entre o OD 84 (LP (2) S26) e o OA 483
(BD*(2) C30-N31), com 47,22 Kcal/mol; entre o OD 82 (LP (3) S25)
e o OA 483 (BD*(2) C30-N31), com 51,19 Kcal/mol; entre o OD 95
(LP (3) S29) e o OA 479 (BD*(1) S26-Fe28), com 64,12 Kcal/mol;
entre o OD 81 (LP (2) S25) e o OA 481 (BD*(1) S27-Fe28), com
67,48 Kcal/mol; e entre o OD 96 (LP (1) N32) e o OA 475 (BD*(2)
C24-S27), com 107,23 Kcal/mol.
A análise preliminar das propriedades ADME/TOX mostra que o composto
apresenta elevada absorção intestinal humana (HIA% de 99,742524) (ALLIANCE,
2016). A permeabilidade celular in vitro Caco-2 de 57,9825 nm/s indica boa
absorção oral e intestinal (OBRIGER et al, 2016). A permeabilidade dérmica
log Kp de -4,15285 (cm/h) sugere baixa absorção cutânea (MORGANTI et al,
2001). A penetração da barreira hemato-encefálica com valor de 4,12053
indica ser um composto ativo no sistema nervoso central (MA, 2005).
Obtivemos ligação de 100.00% com as proteínas plasmáticas, o que prevê
limitação para a distribuição do composto para tecidos e meios
intracelulares (SHARGEL, 2005). O composto também se apresenta como possível
inibidor da glicoproteína-P, o que o torna possível candidato para o
desenvolvimento de terapias anticâncer (AMIN, 2013). Os testes in silico
indicam, ainda, que o composto é mutagênico, porém a predição carcinogênica
em ratos e camundongos foi inconclusiva. Os resultados indicam que as
qualidades para o desenvolvimento racional de um fármaco com o composto
demandam cautela (DIXIT & KUMAR, 2018), embora sugiram possíveis aplicações
antineoplásicas, como já ocorre com outros ditiocarbamatos. Pesquisas
adicionais são necessárias para avaliar melhor o potencial farmacológico do
composto.
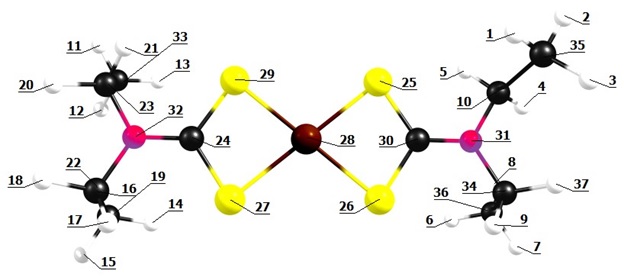
Estrutura geométrica otimizada para o complexo [Fe(DDTC)2], obtida pelo procedimento B3LYP/6- 311G(d, p).
Conclusões
No presente trabalho, descrevemos os orbitais naturais de ligação para o composto de coordenação dietilditiocarbamato de Fe(II): [Fe(DDTC) 2] e obtivemos, computacionalmente, informações a respeito da constituição dos orbitais moleculares na formação das ligações centrais que o complexo apresenta: Fe-S, N=C, C-S e C-C. Para a geometria do composto, obtivemos concordância entre os dados calculados e os dados experimentais obtidos pelos autores que sintetizaram o composto originalmente (ILEPERUMA & FELTHAM, 1975). Informamos também a polarização das ligações em porcentagem sobre os átomos participantes, bem como a ocupação eletrônica das ligações. Os processos de transferência de carga foram analisados no contexto das ligações químicas e também entre os diferentes tipos de orbitais atômicos, inclusive os mais afastados. Embora diversos compostos ditiocarbamatos apresentem aplicações industriais e biomédicas, a literatura ainda é incipiente no que diz respeito a aplicações específicas para o [Fe(DDTC)2]. A carência de aplicações justifica nosso estudo teórico para avaliar as propriedades estruturais e eletrônicas do composto. É importante notar que ditiocarbamatos de Ferro (II) têm sido utilizados para detectar óxido nítrico em aplicações biomédicas e também são potenciais candidatos para terapias antitumorais. Realizamos, portanto, uma avaliação in silico das propriedades farmacocinéticas e toxicológicas do [Fe(DDTC)2]. Os resultados sugerem boa absorção intestinal e por via oral, potencial de atividade no sistema nervoso central, alta afinidade com proteínas plasmáticas (o que sugere possíveis limitações na distribuição do fármaco no organismo) e capacidade de inibição da glicoproteína-P, o que abre a possibilidade de desenvolvimento de aplicações antitumorais. Os dados não foram conclusivos quanto à carcinogenicidade do composto e indicam potencial mutagênico, o que indica a necessidade de cautela em aplicações farmacológicas.
Agradecimentos
O presente trabalho foi realizado com apoio da Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES).
Referências
ALLIANCE, D. Estudo químico, predições in silico das propriedades ADME/TOX e atividade larvicida do óleo essencial da raiz Philodendron deflexum Poepp. Ex Schott SOBRE Aedes egypti Linneu E Anopheles albirtasis s.l. Dissertação (mestrado em ciências da saúde). Unifap. Macapá. 2016.
AMIN, M. L. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights, v. 2013, n. 7, p. 27–34, 2013.
ANDRIENKO, G. A. Chemcraft Version 1.8 (build 595b), 2019.
AYODELE, T. O. & AJIBADE, P. A. Dithiocarbamates: Challenges, Control, and Approaches to Excellent Yield, Characterization, and Their Biological Applications. Bioinorganic Chemistry and Applications, v. 2019, ID 8260496, 2019.
COSTA, A.C.; ONDAR, G.F.; VERSIANE, O.; RAMOS, J.M.; SANTOS, T.G.; MARTIN, A.A.; RANIERO, L.; BUSSI, G.G.A. & TÉLLEZ SOTO, C.A. DFT: B3LYP/6-311G (d, p) vibrational analysis of bis-(diethyldithiocarbamate)zinc (II) and natural bond orbitals. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, v. 105, p. 251-258, 2013.
COSTA, A.C.; RAMOS, J.M.; TÉLLEZ SOTO, C.A.; MARTIN, A.A.; RANIERO, L.; ONDAR, G.F.; VERSIANE, O. & MORAES, L.S. Fourier Transform Infrared and Raman spectra, DFT: B3LYP/6-311G(d, p) calculations and structural properties of bis(diethyldithiocarbamate)copper(II). Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, v. 105, p. 259-266, 2013b.
CVEK, B. & DVORAK, Z. Targeting of Nuclear Factor-κB and Proteasome by Dithiocarbamate Complexes with Metals. Current Pharmaceutical Design, v. 13, n. 30, p. 3155-3167, 2007.
DIXIT, M., KUMAR, A. Mutagenicity: Assays and Aplications. India: Academic Press, 2018.
GAUSSIAN 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian, Inc., Wallingford CT, 2013.
GLENDENING, E. D. ; LANDIS, C. R. ; WEINHOLD, F. NBO 6.0: Natural bond orbital analysis program. Journal of Computational Chemistry, v. 34, n. 16, p. 1429-1437, 2013.
ILEPERUMA, O. A. & FELTHAM, R. D. Crystal and Molecular Structure of Iron(II) Dis(diethyldithiocarbamate). Inorganic Chemistry, v. 14, n. 12, p. 3042-3045, 1975.
KLESCHYOV, A. L. & MÜNZEL, T. Advanced Spin Trapping of Vascular Nitric Oxide Using Colloid Iron Diethyldithiocarbamate. In: CADENAS, E. & PACKER, L. (Eds.). Methods in Enzymology Nitric Ocide, Part D. Burlington: Elsevier Science, 2002. p. 42-51.
KOCH, W. & HOLTHAUSEN, M. C. A Chemist's Guide to Density Functional Theory. Weinheim: Wiley, 2001.
LANDIS, C. R. & WEINHOLD, F. 18‐electron rule and the 3c/4e hyperbonding saturation limit. Journal of Computational Chemistry, v. 37, n. 2, p. 237-241, 2016.
LEE, C. ; YANG, W. & PARR, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B, v. 37, n. 2, p. 785-789, 1988.
LÖWDIN, P. -O. Quantum Theory of Many-Particle Systems. I. Physical Interpretations by Means of Density Matrices, Natural Spin-Orbitals, and Convergence Problems in the Method of Configurational Interaction. Physical Review, v. 97, n. 6, p. 1474-1489, 1955.
MA, X., CHEN, C., YANG, J. Predictive model of blood-brain barrier penetration of organic compounds, Acta Pharmacologica Sinica. 2005, v. 26, p. 500-512.
MANAV, N. ; MISHRA, A. K. & KAUSHIK, N. K. In vitro antitumour and antibacterial studies of some Pt(IV) dithiocarbamate complexes. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, vol. 65, no. 1, p. 32-35, 2006.
MARSMAN, M. ; PAIER, J. ; STROPPA, A. & KRESSE, G. Hybrid functionals applied to extended systems. Journal of Physics: Condensed Matter, v. 20, n. 6, 064201, 2008.
MENEZES, D. C. & DE LIMA, G. M. Aspectos gerais da química dos ditiocarbamatos e de seus complexos metálicos e interações dessas espécies químicas com importantes enzimas - uma breve revisão. Química Nova, v. 44, n. 8, p. 1012-1019, 2021.
MORGANTI, P.; RUOCCO, E.; WOLF, R. & RUOCCO, V. Percutaneous absorption and delivery systems. Clinical Dermatology, v. 19, p. 489-501, 2001.
NEVES, A. P.; VARGAS, M. D.; TÉLLEZ SOTO, C. A.; RAMOS, J. M.; VISENTIN, L. do C.; PINHEIRO, C. B.; MANGRICH, A. S. & REZENDE, E. I. P. de. Novel zinc(II) and copper(II) complexes of a Mannich base derived from lawsone: Synthesis, single crystal X-ray analysis, ab initio density functional theory calculations and vibrational analysis. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, v. 94, p. 152–163, 2012.
OBRINGER, C.; MANWARING, J.; GOEBEL, C.; HEWITT, N. J. & ROTHE, H. Suitability of the in vitro Caco-2 assay to predict the oral absorption of aromatic amine hair dyes. Toxicology in vitro, v. 32, p. 1-7, 2016.
ODULARU, A. T. & AJIBADE, P. A. Dithiocarbamates: Challenges, Control, and Approaches to Excellent Yield, Characterization, and Their Biological Applications. Bioinorganic Chemistry and Applications, vol. 2019, ID 8260496, 2019.
PREADMET. ADME/TOX Prediction. Disponível em: https://preadmet.bmdrc.kr/. Copyright 2005-2017. Acesso em: 09 set 2021.
SHARGEL, L. Applied Biopharmaceutics & Pharmacokinetics. New York: McGraw-Hill, 2005.
TÉLLEZ, C. A.; COSTA JR., A. C.; VERSIANE, O.; LEMMA, T.; MACHADO, N. C. F.; MONDRAGÓN, M. A. & MARTIN, A. A. Surface enhanced Raman scattering, natural bond orbitals and Mulliken atomic charge distribution in the normal modes of diethyldithiocarbamate cadmium (II) complex, [Cd(DDTC)2]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, vol. 146, p. 192-203, 2016.
TÉLLEZ, C. A.; COSTA JR., A; MONDRAGÓN, M. A.; FERREIRA, G. B.; VERSIANE, O.; RANGEL, J. L.; LIMA, G. M. & MARTIN, A. A. Molecular structure, natural bond analysis, vibrational and electronic spectra, surface enhanced Raman scattering and Mulliken atomic charges of the normal modes of [Mn(DDTC)2] complex. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, vol. 169, p. 95-107, 2016.
TOPPING, R. J. & JONES, M. M. Optimal dithiocarbamate structure for immunomodulator action. Medical Hypotheses, vol. 27, no. 1, p. 55-57, 1988.
UCHIDE, N. & OHYAMA, K. Antiviral function of pyrrolidine dithiocarbamate against influenza virus: the inhibition of viral gene replication and transcription. Journal of Antimicrobial Chemotherapy, vol. 52, no. 1, p. 8-10, 2003.
VANIN, A. F.; MORDVINTCEV, P. I.; HAUSCHILDT, S. & MÜLSCH, A. The relationship between l-arginine-dependent nitric oxide synthesis, nitrite release and dinitrosyl-iron complex formation by activated macrophages. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, v. 1177, n. 1, p. 37-42, 1993.
VEKTARIENE, A. The Transition Metal to Ligand Bonding Nature: A Quantum Chemical Study of pi-Allyl-Ruthenacycle Molecule. Lithuanian Journal of Physics, v. 58, n. 3, p. 232-245, 2018.
WEINHOLD, F. & LANDIS, C. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective. Cambridge: Cambridge University Press, 2005.
WEINHOLD, F. ; LANDIS, C. R. & GLENDENING, E. D. What is NBO analysis and how is it useful? International Reviews in Physical Chemistry, v. 35, n. 3, p. 399-440, 2016.
YOSHIKAWA, Y. ; ADACHI, Y. & SAKURAI, H. A new type of orally active anti-diabetic Zn(II)-dithiocarbamate complex. Life Sciences, vol. 80, no. 8, p. 759-766, 2007.