
SENSOR ELETROQUÍMICO À BASE DE FTALOCIANINA DE COBALTO PARA ANÁLISE SIMULTÂNEA DE ACENAFTENO E NAFTALENO EM AMOSTRAS DE POSTOS DE COMBUSTÍVEIS
- Home
- Trabalhos
ÁREA
Química Ambiental
Autores
Paula Mota Ferreira, A. (UFMA (LPQA/BIONORTE)) ; de Sousa Oliveira, E. (UFMA (LPQA/ PPGQUIM)) ; de Andréa Pinheiro, H. (UFMA (LPQA/BIONORTE)) ; Karla Castro Sousa, J. (UFMA (LPQA/ PPGQUIM)) ; Pereira Marques, E. (UFMA (LPQA/ PPGQUIM)) ; Lopes Brandes Marques, A. (UFMA (LPQA/ PPGQUIM))
RESUMO
Este trabalho propõe um método alternativo para a determinação dos HPAs Acenafteno (ACE) e Naftaleno (NAF) em meio aquoso com um eletrodo de carbono vítreo modificado por ftalocianina de cobalto (ECV/CoPc) utilizando as técnicas voltametria cíclica (VC) e voltametria de pulso diferencial (VPD). Após a otimização dos parâmetros, a oxidação do ACE e do NAF ocorreram em 1,0 e 1,3V (Ag/AgCl), respectivamente. Obteve-se os seguintes resultados para o ACE: LD = 2,85x10-9mol L-1, precisão com CV = 3%, exatidão com recuperação = 99,9%; para o NAF: LD = 3,01x10-9 mol L-1, CV = 4,8%, recuperação = 100,5%. Esses resultados indicam que o procedimento proposto se caracteriza como uma boa alternativa para a análise de ACE e NAF em água subterrânea de posto de combustível.
Palavras Chaves
HPA’s; Águas subterrâneas; Posto de combustível
Introdução
A perda contínua de qualidade das águas superficiais, devido aos intensos processos de degradação ambiental ocorridos nos últimos anos tem elevado os custos de tratamento para a potabilização deste recurso, fazendo com que as águas subterrâneas sejam vistas como potenciais fontes de abastecimento de água de boa qualidade e de baixo custo (GEBARA, et al, 2013). As principais fontes potenciais de contaminação das águas subterrâneas são: os lixões; aterros mal operados; acidentes com substâncias tóxicas; atividades inadequadas de armazenamento, manuseio e descarte de matérias primas, produtos, efluentes e resíduos em atividades industriais, como indústrias químicas, petroquímicas podem provocar problemas de salinização ou aumentar a lixiviação de contaminantes para a água subterrânea (FOSTER, et al, 2002). Devido ao aumento de áreas impactadas por hidrocarbonetos surge a necessidade de quantificar esses contaminantes em amostras de água subterrânea, tendo em vista a importância que ela possui como reserva de água potável. Os hidrocarbonetos poliaromáticos são substâncias perigosas, em razão da sua toxicidade, pois possuem estrutura química complexa e que possuem a capacidade de se biocumalarem e aumentarem sua concentração na cadeia alimentar (VERHART,2017). Entre os diversos hidrocarbonetos poliaromáticos, no Brasil a Portaria de Consolidação nº 05 do MS de 28 de outubro de 2017, define o padrão de potabilidade de água para consumo humano, preconiza somente o benzo (a) pireno, com valor máximo permitido de 0,7 mg L-1 como uma substância de análise semestral obrigatória. Não há um mapeamento de todas as áreas contaminadas do Brasil, bem como o conhecimento de outros contaminantes. Os postos distribuidores de combustíveis se constituem atualmente uma das maiores preocupações, pois se encontram bastante dispersos e a quantidade de combustível estocada em cada um deles, pode se derramar no solo, e isso pode ser suficiente para inviabilizar o consumo de milhões de metros cúbicos de água subterrânea (WANG, et. Al, 2013), portanto, é de grande importância o monitoramento de HPA’s em água subterrânea. As fontes naturais mais relevantes são: queimadas de florestas; atividades vulcânicas e decomposição de material biológico, enquanto as fontes antropogênicas mais comuns são as provenientes da combustão do carvão , gás natural ,derivados de petróleo e madeira (para geração de energia e aquecimento) , combustão de derivados de petróleo (para movimentação de embarcações, veículos terrestres e aviões) , atividades industriais (que utilizam derivados de combustíveis fósseis como matéria prima) e queimadas intencionais (de áreas de cobertura vegetal) (WU, 2017;GEBARA, et al, 2013;TOVIDE, 2014; WU, 2017;) Entre as fontes antropogênicas, uma grande preocupação atual é a contaminação causada pelos postos de combustíveis. Nos postos de revenda ou distribuidoras, todos os combustíveis são armazenados em tanques subterrâneos e a não substituição desses tanques de armazenamento é um dos principais motivos que levam a contaminação da água subterrânea (QIAO, 2013). Os tanques subterrâneos de armazenamento de óleo diesel são geralmente instalados próximos aos tanques de etanol. Em caso de derramamento ou vazamento, grandes proporções de óleo diesel poderão entrar em contato com o álcool e as possíveis interações entre os constituintes do diesel e do etanol podem afetar o comportamento destes na superfície (OLIVEIRA; LOUREIRO,1998). Não há um mapeamento de todas as áreas contaminadas do Brasil, bem como o conhecimento de outros contaminantes. Os postos distribuidores de combustíveis se constituem atualmente uma das maiores preocupações, pois se encontram bastante dispersos e a quantidade de combustível estocada em cada um deles, pode se derramar no solo, e isso pode ser suficiente para inviabilizar o consumo de milhões de metros cúbicos de água subterrânea (WANG, et. Al, 2013), portanto, é de grande importância o monitoramento de HPA’s em água subterrânea. Atualmente, o método mais utilizado para detecção de HPA’s são os cromatográficos. Contudo, os métodos voltamétricos, são uma alternativa interessante para determinação de HPA’s, pois apresentam menor manipulação de amostras e menor custo de análise quando comparadas as técnicas cromatográficas (SIODA, 2004; SIODA, 2008). Esse trabalho, desenvolveu uma metodologia alternativa para a determinação de hidrocarbonetos poliaromáticos (ACE e NAF), utilizando um eletrodo de carbono vítreo quimicamente modificado com ftalocianina de cobalto usando a técnica de voltametria de pulso diferencial.
Material e métodos
Todos os reagentes usados no trabalho eram de grau analítico e usados sem purificação adicional. As soluções foram preparadas com água deionizada Milli-Q com resistividade ≥ 18 MΩ cm-1. Para a obtenção de uma superfície ativa e reprodutível, o ECV foi submetido a um polimento manual em suspensão de alumina com granulação 0,3 µm. Em seguida, o eletrodo foi lavado com água purificada, e, posteriormente, levado para um sistema de banho ultrassom por três minutos, para a remoção de impurezas da superfície do eletrodo. Todos os reagentes utilizados neste trabalho foram de grau analítico e preparados com água purificada num sistema Milli-Q da Millipore. Durante os experimentos, toda vidraria utilizada passou por um procedimento de limpeza para assegurar a ausência de quaisquer resíduos orgânicos ou metálicos que pudessem interferir nas análises. As amostras de água foram coletadas em um posto de distribuição de combustíveis localizados na região central de São Luís-MA. Para a coleta, foram utilizados frascos de vidro âmbar previamente lavados com detergente comum em água corrente e posteriormente em solução de Extran (5%, neutro) por aproximadamente 24 horas. Os frascos foram enxaguados em água ultrapura e secos em estufa a 100 ° C por 24 horas. As amostras foram transportadas em banho de gelo até o laboratório e armazenadas a aproximadamente 4 ° C por um período máximo de 20 dias para os experimentos A solução de CoPc foi preparada pela dissolução de 0,0028 g em 4500µL de metanol e 500µL de nafion, obtendo-se a concentração de 1x10-3 mol L-1. A solução de acenafteno foi preparada em um mix de 25% de acetonitrila e 75% de água e a solução de NAF foi preparada em um mix de 25% de acetonitrila e 75% de água (Merck). A solução Britton Robinson foi preparada pela mistura 50 mL de uma solução H3BO3 0,2 mol L-1 com 50 mL de CH3COOH 0,2 mol L-1 e 50mL de H3PO4 0,2 mol L-1 seguido pela adição de NaOH 1 mol L-1 de modo a obter o valor do pH desejado (medido no pH metro). Solução Alcalina de Permanganato de Potássio: foram dissolvidos 3,0 g de KOH (Merck) em 1 (um) litro de água e adicionou-se 1,5 g de KMNO4 (Merck). Após a remoção das partículas no banho de ultrassom o método para a modificação do ECV foi o de adsorção irreversível. Primeiramente foi feita uma solução metanólica de CoPc na concentração de 1x0-3 mol L-1, contendo 5% de nafion. Em seguida, foi pingado no eletrodo uma quantidade de 10µL em sua superfície, esperou-se sua secagem em um dessecador a vácuo e logo após mergulhado na solução eletrolítica e levado ao equipamento potenciostato/galvanostato PGSTAT 302 da Metrohm-Autollab, utilizado para as medidas eletroquímicas.
Resultado e discussão
Experimentos preliminares de voltametria cíclica foram realizados a fim de se
observar os processos de oxidação de ACE (C12H10) e NAF (C10H8) sobre a
superfície do ECV não modificado ou modificado com CoPc.
Estudou-se a oxidação do ACE e NAF com o eletrodo quimicamente modificado
(ECV/CoPc) (voltamograma C) e com o eletrodo não modificado (ECV) (voltamograma
B). Um voltamograma com o eletrodo ECV/CoPc também foi obtido (voltamograma A)
na ausência dos analitos, o qual não apresentou picos voltamétricos.
Algumas otimizações foram feitas para a análise do ACE e NAF por voltametria
cíclica. Os melhores resultados foram os seguintes: (1) Eletrólito suporte;
Tampão BR, Tampão Macllvaine, Tampão Sörenser e Ácido de Sulfúrico (Melhor
resultado: Tampão BR); (2) pH: pH: 2,0, 3,0,4,0,5,0,6,0,7,0 e 8,0 (Melhor
resultado: pH 2
Após a realização dos experimentos, para determinação individual dos analitos
ACE e NAF, realizou-se o estudo para a determinação simultânea dos analitos por
voltametria de pulso diferencial (VPD), utilizando-se ECV/CoPc. Portanto, alguns
parâmetros foram otimizados para a técnica de VPD, que foi escolhida para
realizar o estudo simultâneo, pois foi a técnica que demonstrou melhor
sensibilidade para ambos os analitos, determinados de forma individual. As
otimizações realizadas foram: amplitude e tempo de pulso.
Os melhores resultados foram os seguintes: (1) Amplitude: 10, 20, 30, 40, 50,
60, 70, 80, 90 e 100mV (Melhor resultado: 70 mV); (2) Tempo de Pulso: 200, 300,
400 e 500 ms (Melhor resultado: 200 ms). Construiu-se curvas analíticas para a
determinação simultânea dos analitos.
Na detecção simultânea, podemos observar que houve uma dependência linear entre
a concentração dos analitos (Figura 2) e a corrente de pico e obtemos as
seguintes equações da reta para ACE de Ip = 3,184 + 19.480 [ACE], R = 0,980 e
para NAF foi Ip = 3.320 + 18.963 [NAF], R = 0,988. Obtivemos como LD para ACE um
valor de 2,85x10 -9 mol L-1 e para NAF 3,01x10-9 mol L-1, esses valores obtidos
demonstraram uma boa sensibilidade apara análise simultânea.
A aplicação do procedimento otimizado em amostra real foi feita através do
procedimento de enriquecimento da amostra, usando o método de adição padrão.
Neste procedimento, 4 mL da amostra diretamente na célula eletroquímica contendo
6 mL de solução tampão BR 0,2 mol L-1 (pH 2,0). Os valores contidos na amostra,
foram calculados utilizando-se a extrapolação da reta do gráfico de adição
padrão, conforme apresentados equações abaixo, através das quais, as
concentrações dos analitos foram determinadas na amostra real.
Ip= 0,07363 + 6,22 [ACE], r = 0,999 (VPD)
Ip= 0,65 + 14,72[NAF], r = 0,998 (VPD)
As concentrações encontradas na amostra real foram de 0,012 µmol L-1 (1,2x10-8
mol L-1) para ACE e 0,044 µmol L-1 (4,4 x10-8 mol L-1) para NAF.
Os resultados obtidos com o sensor proposto apresentaram boa faixa sensibilidade
e baixo limite de detecção para ACE e NAF, considerando a matriz em estudo.
A RESOLUÇÃO CONAMA 420/2009 mostra a concentração mínima aceitável do HPA NAF
(128,17 g por mol) como sendo 0,12 mg L-1 e não existe valor de referência para
o ACE (154,2 g por mol).
Os resultados obtidos para NAF (0,0056 mg L-1) e ACE (0,0018 mg L-1) indicam que
a água subterrânea se encontra em condições de potabilidade.
A sensibilidade do método, expressa pelo seu limite de detecção (LD) do ACE e
NAF de 2,85 x 10-9 e 3,01 x 10-9 mol L-1 respectivamente, frente à norma vigente
(CONAMA: 4,37x10-2 mol L-1) indica que o procedimento proposto pode ser
considerado como uma importante alternativa para controle e monitoramento da
qualidade das águas subterrâneas.
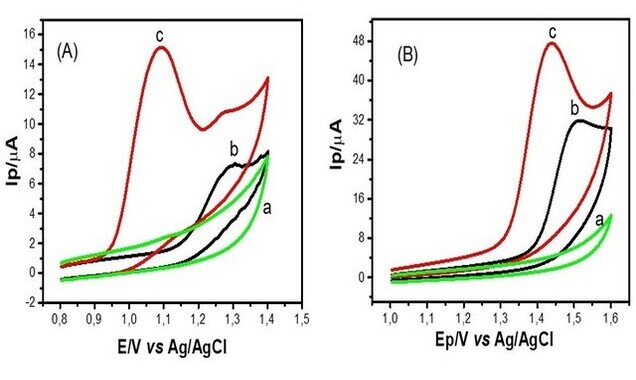
Voltamogramas cíclicos: (a) (ECV/CoPc) branco, (b) ECV com os analitos ACE e NAF (c) ECV/CoPc com os analitos ACE e NAF, v = 0,05 V s-1.
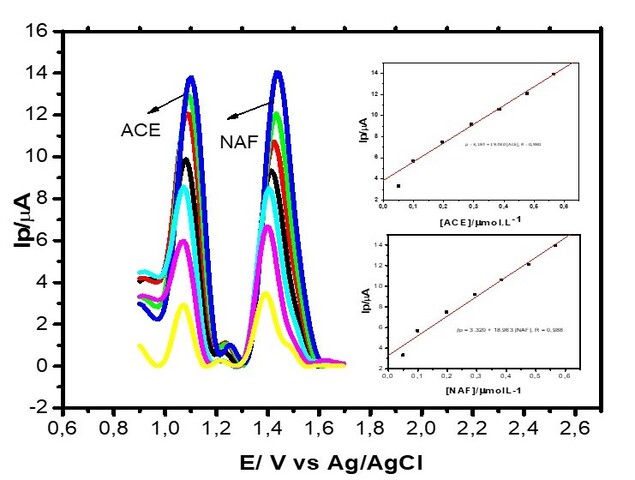
VPD para o ECV/CoPc com [ACE e NAF] de 4,97x10-8 a 5,66x10-7 molL-1 em Tampão BR pH 2, v = 0,025 Vs- 1. Inserção: curvas Analíticas de ACE e NAF
Conclusões
Os estudos realizados através da técnica de voltametria cíclica mostram que a resposta do eletrodo de carbono vítreo modificado com a ftalocianina de cobalto (CoPc) é melhor, do ponto de vista eletroanalítico, quando comparado ao eletrodo não modificado. O uso do eletrodo quimicamente modificado com CoPc proporcionou um aumento significativo nas correntes de pico de oxidação dos analitos, em comparação com o eletrodo não modificado, indicando maior sensibilidade eletroanalítica e melhor atividade eletrocatalítica deste sensor para análise de ACE e NAF. A investigação do efeito do pH sobre o sinal analítico do sensor mostrou máxima sensibilidade na determinação do ACE e NAF em pH 2,0, sendo o tampão BR (0,2 mol L-1) o eletrólito que mostrou maior sensibilidade para ambos os analitos. A concentração dos analitos encontrada para NAF (0,0056 mg L-1) e ACE (0,0018 mg L-1) indicam que a água subterrânea se encontra em condições de potabilidade. Os limites de detecção encontrados para ACE (2,85 x 10-9 mol L-1) e NAF (3,01 x 10-9 mol L-1), indicam que o procedimento proposto pode ser considerado como uma alternativa promissora para avaliação da qualidade das águas subterrâneas.
Agradecimentos
CAPES (PROCAD-AM / SCBA 450 88887.200615 / 2018-00], CNPq [PQ 2017, Proc. 310664 / 2017-9], e ANP [1.029 / 2016-ANP-007.639].
Referências
FOSTER, S. HIRATA, R.; GOMES, D.; D'ELIA, M.; PARIS, M. Groundwater quality protection: a guide for water service companies, municipal authorities and environment agencies. The World Bank, n°1, 16-17, 2002.
GEBARA, S. S; RÉ-POPPI, N.; NASCIMENTO, A. L. C. S.; RAPOSO JUNIOR, J. L. Methods for analysis of PAH and BTEX in groundwater from gas stations: a case study in Campo Grande, MS, Brazil. Química Nova, n°7, 1030-1037, 2013.
OLIVEIRA, L. I.; LOUREIRO, C. O. Contaminação de aquíferos por combustíveis orgânicos em Belo Horizonte: Avaliação preliminar. Águas Subterrâneas, n°1, 2-5, 1998.
QIAO, M.; QI, W.; LIU, H; QU, J. Simultaneous determination of typical substituted and parent polycyclic aromatic hydrocarbons in water and solid matrix by gas chromatography–mass spectrometry. Journal of Chromatography A, n°1, 129-136, 2013.
SIODA, R. E.; FRANKOWSKA, B. Electro-oxidative reactions of naphthalene and alkyl-derivatives, ANNALES… Universitatis Mariae Curie-Sklo Dowska, n°14, 154-165, Lublin-Polonia, 2004.
SIODA, R. E.; FRANKOWSKA, B. Voltammetric oxidation of naphthalene derivatives. Journal of Electroanalytical chemistry, n°1, 147-150, 2008.
TOVIDE, O.; JAHEED, N.; MOHAMED, N.; NXUSANI, E.; SUNDAY, C. E.; TSEGAYE, A.; AJAYI, R. F.; NJOMO, N.; MAKELANE, H.; BILIBANA, M.; BAKER, P. G.; WILLIAMS, A.; VILAKAZI, S.; TSHIKHUDO, R.; IWUOHA, E. I. Graphenated polyaniline-doped tungsten oxide nanocomposite sensor for real time determination of phenanthrene. Electrochimica Acta, n°14, 138-148, 2014.
VERHAERT, V.; D'HOLLANDER W.; COVACI, A.; VLOK, W.; WEPENER, V.; ADDO-BEDIAKO, A.; JOOSTE, A.; TEUCHIES, J.; BLUST, R.; BERVOETS, L. Persistent organic pollutants in the Olifants River Basin, 47 South Africa: Bioaccumulation and trophic transfer through a subtropical aquatic food web. Science of the Total Environment, n°15, 792–806, 2017.
WANG, X.-T.; MIAO, Y.; ZHANG, Y.; LIA, Y.-C.; WU, M.-H.; YU, G. Polycyclic aromatic hydrocarbons (PAHs) in urban soils of the megacity Shanghai: occurrence, source apportionment and potential human health risk. Science of the Total Environment, n°1, 80-89, 2013.
WU, R.; LI, N.; SHU, R.; AN, N.; YI, F.; YANG, W.; LI, C. Determination of Polycyclic Aromatic Hydrocarbons in Mosses by Ultrasonic-Assisted Extraction and Gas Chromatography–Tandem Mass Spectrometry. Analytical Letters, n°1, 243-257, 2017.