Autores
da Silva Ramos, R. (UNIVERSIDADE DO ESTADO DO AMAPÁ) ; Fauro de Araujo, I. (UNIVERSIDADE FEDERAL DO AMAPÁ) ; dos Santos, I.V.F. (UNIVERSIDADE FEDERAL DO AMAPÁ) ; da Silva Costa, J. (UNIVERSIDADE DO ESTADO DO AMAPÁ) ; Rodrigues dos Santos, C.B. (UNIVERSIDADE FEDERAL DO AMAPÁ)
Resumo
O estudo teve como objetivo identificação de potenciais inibidores e
investigação do mecanismo de ação no receptor SARS‑CoV‑2 (Spike/ACE2) usando
estudo de docking molecular. As moléculas promissoras foram provenientes de
triagem virtual hierárquica a partir da hidroxicloroquina e análogos de 1,2,4,5
tetraoxanos. O receptor, os ligantes de referencias e as moléculas promissoras
foram preparados (otimizadas, minização de energia e adicionaodos os H) usandos
programas específicos para as simuçações de docking molecular. Os protocolos de
docking foram validados por método de cálculo do RMSD, os valores de afinidade e
interações avaliadas. O ligante MolPort-005-060-605 apresentou o melhor
resultado de afinidade de ligação (ΔG) de -8,1 kcal/mol comparado ao controle
usado.
Palavras chaves
Covid; Pandemia ; Docking molecular
Introdução
Uma epidemia começou em dezembro de 2019 em Wuhan, China, nas quais as pessoas
infectadas sofriam de sintomas semelhantes aos da pneumonia, que mais tarde se
espalhou por todo o mundo. Descobriu-se que a causa principal da infecção era um
novo vírus que apresenta semelhanças estruturais com Coronavírus relacionados à
Síndrome Respiratória Aguda Grave, portanto, denominado de SARS-CoV-2 (Zhu et
al., 2020; Zhou et al., 2020).
De acordo com os dados da OMS (https://www.who.int/emergencies/diseases/novel-
coronavirus-2019/situation-reports), atualmente (a partir de 19 de setembro de
2021), o número de casos semanais de COVID-19 e mortes em todo o mundo continuou
a diminuir esta semana, com mais de 3,6 milhões de casos e pouco menos de 60.000
mortes relatadas entre 13-19 de setembro. Isso traz o número cumulativo de casos
confirmados e mortes em todo o mundo para quase 228 milhões e mais de 4,6
milhões, respectivamente.
Assim, devido a urgência de estratégias de um tratamento eficaz, através do
uso de medicamentos virais relatados na literatura por apresentarem grandes
vantagens, pois a farmacocinética, a farmacodinâmica e os perfis de segurança
desses medicamentos já foram estabelecidos. O reposicionamento de fármaco e a
triagem virtual baseado em ligante, ganharam relevante importância, pois se
espera resultados mais rápidos e com menos investimento para a identificação de
um potente agente antiviral.
Estudos preliminares revelaram a terapia combinada de lopinavir/ritonavir como
um potencial inibidor do vírus. Junto com essas duas drogas, muitas outras
drogas antivirais também foram testadas. Recentemente, foi relatado que os
fármacos antimaláricos cloroquina e a hidroxicloroquina têm certo efeito
curativo sobre COVID-19, no entanto, os fármacos apresentam alerta de
hepatotoxicidade (Purwati et al., 2020; Wang et al., 2020; Das et al., 2021). O
SARS-CoV-2 é um vírus de RNA de fita simples de sentido positivo que depende de
sua proteína Spike (S) para se ligar e entrar nas células-alvo. A proteína S do
vírus se liga ao receptor da enzima conversora de angiotensina 2 (ACE2) da
célula hospedeira, permitindo que as partículas do vírus entrem nas células.
Assim, o bloqueio do receptor ACE2 revela um potencial alvo terapêutico para a
descoberta de drogas para prevenir a transmissibilidade da SARS-CoV-2 (Baysal et
a., 2021; Basu et al., 2021).
A hidroxicloroquina (HCQ) foi usada como molécula pivô, de acordo com Wang et
al (2020). Estudos [in vitro][/in vitro] relatados para HCQ eficaz contra SARS-
CoV-2 em uma Multiplicidade de Infecção (MOI) de 0,01 com uma concentração
efetiva de 50% ([EC][/50]) foi de 4,51 μM em células Vero E6. Todos os MOIs
(0,01, 0,02, 0,2 e 0,8) e EC50 para HCQ (4,51, 4,06, 17,31 e 12,96 μM) foram
satisfatórios.
A modelagem de moléculas bioativas tem sido bastante utilizada na
identificação de novos protótipos com atividade biológica, e essa busca consiste
em pré-selecionar compostos com o auxílio do computador a partir de bancos de
dados virtuais.Logo, o estudo teve como objetivo identificação de potenciais
inibidores e investigação do mecanismo de ação no receptor SARS‑CoV‑2
(Spike/ACE2) usando estudo de docking molecular.
Material e métodos
As moléculas promissoras foram provenientes de triagem virtual hierárquica a
partir da hidroxicloroquina e análogos de 1,2,4,5 tetraoxanos. A atribuição
correta dos estados de protonação/tautomérico da proteína e dos
ligantes é crucial para o modo de ligação e suas previsões de afinidade,
exigindo uma inspeção cuidadosa das estruturas. Nesta pesquisa, os complexos
foram preparados usando o servidor web PDB2QR
(https://server.poissonboltzmann.org/pdb2pqr). A atribuição de protonação e
estados tautoméricos dos ligante foi realizado com o programa Discovery Studio,
enquanto os átomos de hidrogênio da proteína foram adicionados com PROPKA usando
o pH 7.
A estrutura cristalina do domínio de ligação ao receptor Spike do SARS-CoV-2
ligado a ACE2 (organismo Homo sapiens com PDB ID 6M0J (LAN et al., 2020),
resolução de 2.45 Å, elucidada pelo método de difração de raio-X e
foi baixada no Protein Data Bank (PDB) no formato (.pdb) para realizar estudo de
interação e modo de ligação receptor-ligante. O ligante hidroxicloroquina foi
utilizada como controle positivo e todas as moléculas de água e cofatores
retiradas.
As simulçaoes de docking molecuar foram desenvolvidas no servidor DockThor; a
configuração do grid box de cada complexo foi determinado automaticamente de
acordo com o ligante de referência quando disponível: (1) O centro das
coordenadas foi definido como o centro das coordenadas do ligante. (2) O tamanho
da grid foi definido como o maior valor do eixo do ligante, mas com uma
tolerância de 6 Å em cada dimensão. (3) A discretização (ou seja, espaçamento
entre os pontos do grid box) foi definido com o valor padrão de 0,25 Å (SANTOS
et al., 2020).
Os parâmetros padrão do algoritmo usado foram definidos da seguinte forma: (1)
24 corridas de conformações de encaixe, (2) 1000000 avaliações por corrida de
encaixe e (3) população de 750 indivíduos. A qualidade de pontuação do docking
da proteína-ligante foi avaliada com base no RMSD entre o melhor score da pose
de docking.
Resultado e discussão
Dada a grande superfície de contato entre o domínio RDB da Spike e ACE2, para
realizar estudos de docking neste local de ligação, a configuração do grid foi
centrada no Cα do resíduo GLN493 localizado na interface da interação entre
Spike e ACE-2. Segundo Lan et al., (2020) as mensurações de ligação in vitro
mostraram que o SARS-CoV-2 RBD se liga a ACE2 em um baixo intervalo de afinidade
(nanomolar), indicando que o RBD é um componente funcional chave dentro da
subunidade S1 que é responsável pela ligação de SARS-CoV-2 na ACE2. Em
comparação em estudos de alinhamento e mapeamento em suas respectivas sequências
dos resíduos que interagem com ACE2 nos RBDs SARS-CoV-2 e SARS-CoV.
O SARS-CoV-2 RBD tem cinco cadeias torcidas em antiparalelo de β-folha (β1,
β2, β3, β4 e β7) com hélices de conexão curtas e laços que formam o núcleo.
Entre as fitas β4 e β7 no núcleo, há uma inserção estendida contendo as fitas β5
e β6 curtas, hélices α4 e α5 e loops (Figura 8). Esta inserção estendida é o
RBM, que contém a maioria dos resíduos de contato do SARS-CoV-2 que se ligam ao
ACE2. O domínio N-terminal da peptidase de ACE2 tem dois lóbulos, formando o
sítio de ligação do substrato peptídico entre eles.
As poses de docking de todas as moléculas principais mostrou que eles estão
interagindo em uma conformação que as encaixa na bolsa de ligação do RBM. As
poses de docking juntamente com suas respectivas interações são mostradas na
Figura 1.
As poses de docking geradas possibilitou observar que os ligantes interagem
com os resíduos de aminoácidos do sítio ativo da Spike RBD (PDB ID 6M0J) ao
redor da α-hélice entre os resíduos de aminoácidos Tyr449-Tyr505 e compreendidos
na β-folha entre os resíduos de aminoácidos Glu35-Asp39. Nos ligantes, torna-se
possível observar interações hidrofóbicas com a grande maioria dos resíduos em
Leu39, Tyr449, Leu452, Phe490 e Leu492, resultados esses que estão de acordo com
os estudos da literatura (Liu et al., 2020; Faisal et al., 2021) .
No estudo de docking molecular as interações dos potenciais inibidores com os
resíduos de aminoácidos Tyr449, Gln493, Ser494 e Tyr505 na Spike RBD são
semelhantes às relatadas na literatura. O inibidor mais bem avaliado
em termos de afinidade de ligação foi o MolPort-005-060-605 (-8,1 kcal/mol), no
qual as interações foram similares as observadas no controle para os resíduos
Glu35 e Ser494, contribuindo para o aumento da afinidade de ligação. As
interações menos comuns entre os inibidores foram Leu39, Tyr351, Tyr 449,
Phe490, Glu494 e Tyr505, sendo que essas contribuições ajudam na estabilização
no sítio de ligação para inibição da Spike no domínio RBM.
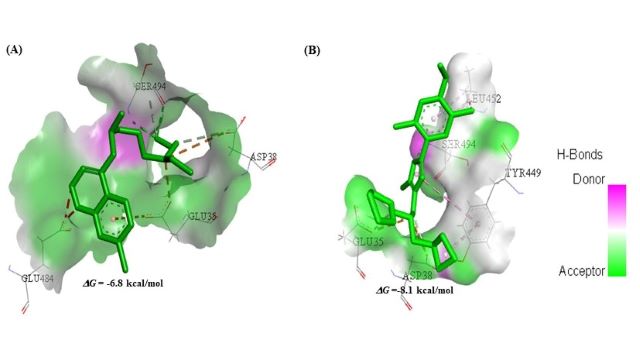
Interações dos ligantes (A) Hidroxicloroquina, (B) MolPort-005-060-605 no sítio ativo da Spike RBD.
Conclusões
A pandemia relacionada COVID-19 é uma luta que ainda precisa ser travada pela
humanidade e além da prevenção por vacinação, a única saída é através da
descoberta de novos medicamentos. Nosso estudo identificou alguns potenciais
candidatos que podem ser usados para a inibição da proteína Spike em COVID-19.
Os estudos de docking molecular confirmaram a ligação das moléculas no sítio de
ligação ACE2, na qual a molécula MolPort-005-060-605 teve melhor valor de
afinidade de ligação para o alvo molécular. Estudos adicionais experimentais (in
vitro e in vivo) necessitam ser realizados para testar os possíveis candidatos,
uma vez que são fáceis de ser sintetizados, e assim, esclarecer melhor o mecanismo
de ação do vírus no organismo humano.
Agradecimentos
A Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) pelo
financiamento do projeto CAPES-EPIDEMIAS. Processo número: 88887.507221/2020-00.
A Universidade Federal do Amapá e a Universidade do Estado do Amapá.
Referências
Zhu H, Wei L, Niu P. The novel coronavirus outbreak in Wuhan, China. Global Health Research and Policy 2020;5. https://doi.org/10.1186/s41256-020-00135-6.
Zhou P, Yang X lou, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. https://doi.org/10.1038/s41586-020-2012-7.
World Health Organization. Global overview (2021, September). Weekly epidemiological update on COVID-19 - 14 September 2021. Weekly epidemiological update on COVID-19 - 14 September 2021 (who.int).
Purwati, Miatmoko A, Nasronudin, Hendrianto E, Karsari D, Dinaryanti A, et al. An in vitro study of dual drug combinations of anti-viral agents, antibiotics, and/or hydroxychloroquine against the SARS-CoV-2 virus isolated from hospitalized patients in Surabaya, Indonesia. PLoS ONE 2021;16.
https://doi.org/10.1371/journal.pone.0252302.
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research 2020;30:269–71. https://doi.org/10.1038/s41422-020-0282-0.
Das S, Sarmah S, Lyndem S, Singha Roy A. An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study. Journal of Biomolecular Structure and Dynamics 2021;39:3347–57. https://doi.org/10.1080/07391102.2020.1763201.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20. https://doi.org/10.1038/s41586-020-2180-5.
Liu Q, Wang Y, Lai-Han Leung E, Yao X. In silico Study of Intrinsic Dynamics of Full-length apo-ACE2 and RBD-ACE2 complex. Computational and Structural Biotechnology Journal 2021. https://doi.org/10.1016/j.csbj.2021.09.032.
Faisal HMN, Katti KS, Katti DR. Binding of SARS-COV-2 (COVID-19) and SARS-COV to human ACE2: Identifying binding sites and consequences on ACE2 stiffness. Chemical Physics 2021;551:111353. https://doi.org/10.1016/j.chemphys.2021.111353
Santos KB, Guedes IA, Karl ALM, Dardenne LE. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein-Peptide Data Set. Journal of Chemical Information and Modeling 2020;60:667–83. https://doi.org/10.1021/acs.jcim.9b00905.














