Autores
de Andrade, K.N. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Martorano, L.H. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Correa, G.S. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Santos Jr, F.M. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Carneiro, J.W.M. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Albuquerque, A.C.F. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Gomes, A.C.C. (INSTITUTO FEDERAL DO RIO DE JANEIRO) ; Fiorot, R.G. (UNIVERSIDADE FEDERAL FLUMINENSE)
Resumo
C-glycosyl flavonoids are an important class of natural products widely known by
their biological activities. Some compounds of that family are found as
rotational isomers, identified by NMR signal duplication. Despite being a well-
known phenomenon, there is no consensus on its origin. In this work, we explored
the structural and solvation effects on the rotational isomerism in five
flavonoids by means of Density Functional Theory. Our results suggest that the
rotational isomerism is associated with the restriction of the C8
–C(glycosyl) bond rotation. Besides, the explicit solvent (DMSO)
interaction seems to be a key point in identifying the NMR duplication
(rotational barrier ΔG‡ ~ 20 kcal mol-1).
Palavras chaves
Atropisomerism; Explicit Solvation; NMR signal duplication
Introdução
C-glycosyl flavonoids are an important class of natural products with relevant
pharmacological activities. (TOENJES, GUSTAFSON, p. 409, 2018) The restricted
rotation around the C(sp3)–C(sp2) bond from
the sugar-aglycone backbones allows rotational isomerism to occur. When the
rotational energy barriers are higher than 20 kcal mol-1, the
conformational isomers are called atropisomers and can be isolated as individual
compounds. The occurrence of this isomerization, which can be identified by
signal duplication in the NMR spectra, is important due to the selective
interaction of each isomer with macromolecules and proteins. (ZHOU et al, p.
443, 2019) However, not all C-glycosyl flavones exhibit rotational isomerism and
the reasons for this phenomenon remain unclear. The rotational isomerism has
been mainly attributed to bulk substituents near to the C(sp
3)–C(sp2), such as in the C7
position, but also to the position and substitution pattern in the B ring. The
solvation environment also seems to have some influence. (ZHOU et al, p. 443,
2019) However, there is no consensus in the literature on which bond rotation
(C6–R1, C8–R3, or B ring rotation,
in bold in Figure 1) would give rise to the rotational isomerism. Herein, we
explored the origin of the rotational isomerism in C-glycosyl flavonoids by
means of the Density Functional Theory. To do so, we selected the following
flavonoids with structural diversity, varying the substituents in R1,
R2, and R3, as well as the position of the B ring:
Isoschaftoside (1), Schaftoside (2), Vitexin (3), Puerarin
(4), and Prunetin (5). We assessed the origin of the isomerism in
terms of substituents (C6, C8, or B ring). Then, the
solvent effect was evaluated from the explicit simulation of solvent molecules –
H2O (1) and DMSO (2-5).
Material e métodos
All the quantum calculations were performed using the DFT B3LYP/6-311++G(d,p)
level at Gaussian 09 package. As hydrogen bonding and non-bonded interactions
play a pivotal role to rotational barriers, we included Grimme’s empirical
dispersion correction (GD3) in all calculations. The nature of the stationary
points was characterized by frequency calculations. Minimum-energy points on the
potential energy surface presented only positive eigenvalues and first-order
saddle points (transition states) were identified presenting only one negative
eigenvalue. Thermodynamic parameters were computed at 298 K and 1 atm. We
regarded solvation effects by two approaches: pure and mixed solvation model.
The first was simulated considering exclusively implicit solvation with the
self-consistent reaction field (SCRF) using the integral equation formalism
variant of the polarizable continuum model (IEFPCM) with water (ε = 78.3) and
dimethylsulfoxide (ε = 46.8) as solvent, depending on the experimental
conditions used to obtain the spectra. For the mixed model, the IEFPCM was
combined with explicit solvation. For that, we performed Molecular Dynamics
simulations at Tinker v. 8.10 software, with OPLS-AA force field. The structures
were placed in cubic boxes with explicit water or dimethylsulfoxide molecules.
The equilibrium phase was simulated in two steps, with NVT and NPT ensembles.
The production phase was performed with a NPT ensemble and consisted of 10,000
steps of 1 fs each, generating a trajectory of 10 ps. From the simulation, one
frame was selected. Due to the high computational cost, all solvent molecules
within a 3.0 Å radius from the solute were selected for the subsequent
optimization at B3LYP/6-31G(d). Then, the electronic energy was refined at
B3LYP-D3/6-311++G(d,p) level.
Resultado e discussão
Figure 1 presents the general structure of the assessed flavonoids. To
understand the substituents effects to the rotational isomerism and identify
which bond is more prone to cause it, we started by computing the energy
barriers related to the C6–R1, C8
–R3, and B ring bond rotation in the pure solvation model.
According to the literature, rotational barriers smaller than 10 kcal
mol-1 are freely rotatable, while barriers ca. 20 kcal
mol-1 would be enough to promote signal duplication in the NMR
spectra and allow the isolation of the atropisomers. (ZHOU et al, p. 443, 2019)
Our results (Table 1, pure solvation) suggest that the rotation around the
C8–R3 bond is more prone to cause the rotational
isomerism, since it has higher energy barrier (ΔG‡ = 13.3–15.1
kcal mol-1) than the alternative C6–R1
and B ring bonds, which seem to be freely rotatable (ΔG‡ < 8
kcal mol-1). Previous works report NMR signal duplication for all the
flavonoids selected herein, except for Puerarin (4). (CHENG et al, p.
8915, 2000). For Schaftoside, the duplication is not unequivocal and the authors
ascribed it as impurity signals (LIU et al, p. 4934309, 2017). A review paper
highlighted that the rotational barriers might be strongly solvent-dependent,
specifically for the DMSO-d6, often used in NMR experiments. (ZHOU et
al, p. 443, 2019). Thus, we employed the mixed microsolvation model to evaluate
the explicit solvent-solute interactions in the rotational isomerism of the
C8–R3 bond. For all cases, these interactions,
established between the DMSO and the glycone portion, Me2SO•••HO–
(R3) increase the rotational energy barriers to values > 17 kcal
mol-1. With the exception of 4, the new energy barriers was
ca. 20 kcal mol-1, being able restrict the bond rotation and
promote the atropisomerism. Thus, our outcomes suggest that solvation effects,
mainly due to hydrogen bonding, is a key point for this phenomenon, in
accordance with the literature. (ZHOU et al, p. 443, 2019) The lowest barrier
was identified for compound 4, compatible with the experimental non-
identification of its rotational isomerism. (CHENG et al, p. 8915, 2000)
Activation strain and energy decomposition analyses are being performed for a
prototype system (Vitexin) to assess the physical reasons to the rotational
barriers and the influence of the solvation.
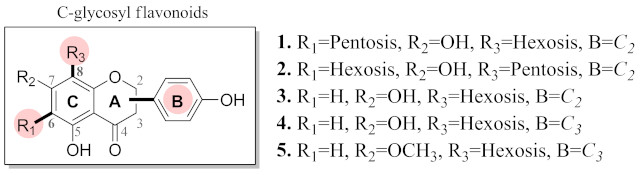
General C-glycosyl flavonoids structure.
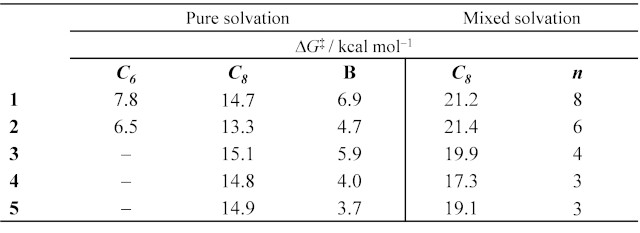
Calculated rotational barriers relative to the most stable conformer using pure and mixed solvation models: H2O (1) and DMSO (2-5).
Conclusões
The C-glycosyl flavonoids are an important class of natural products that exhibit
rotational isomerism. Herein we identified which bond is more prone to cause it
(C6–R1, C8–R3, and B
ring), and assessed how solvation affects this phenomenon. According to our
evaluations, the C8–R3 bond rotation is causing the
rotational isomerism, but the intrinsic substituent effect is not sufficient to
promote restriction in bond rotation. The simulation of the explicit solvent
molecules showed that the solute-solvent interactions play a pivotal role in the
bond rotational energy barrier increase.
Agradecimentos
To CAPES, CNPq and FAPERJ. We also thanks to the Núcleo Avançado de Computação de
Alto Desempenho (Project ID a20006).
Referências
CHENG, G.; BAI, Y.; ZHAO, Y.; LIU, Y.; TU, G.; MA, L.; LIAO, N.; XU, X. [b]Tetrahedron[/b] No 56, p. 8915 2000. LIU J.; LIU Y.; DAI Z.; HE L.; MA, S.[b]Journal of Analytical Methods in Chemistry[/b] No 4934309, 2017. TOENJES, S. T.; GUSTAFSON J. L.; [b]Future Medicinal Chemistry[/b], No 4, 409-422, 2018. ZHOU, G.; YAN, R.; WANG, X.; LI, S.; LIN, J.; LIU, J.; ZHAO, Z.[b]Phytochemistry Reviews[/b], No 18, 443-461, 2019.














