Autores
Raffaeli, N.M. (UFF) ; de Andrade, K.N. (UFF) ; Goetze Fiorot, R. (UFF)
Resumo
Fotointerruptores moleculares são substâncias que a partir de um estímulo
luminoso são convertidas em estados estáveis com propriedades distintas.
Akamptisomeria é um novo tipo fundamental de isomeria baseado na inversão de
ângulo de ligação (IAL) de uma ponte tensionada, como (N)2FB–O–
BF(N)2ancorada em compostos porfirínicos.Nesse processo, a
planaridade dos sistemas é afetada,o que influencia a conjugação dos elétrons π,
tornando tais compostos fotointerruptores em potencial. Avaliou-se como
alterações estruturais afetam a IAL e as propriedades ópticas das porfirinas
utilizando DFT e TD-DFT. Concluiu-se que todas as espécies foram calculadas como
akamptisoméricas que absorvem na região do UV/Vis, porém são inviabilizadas como
fotointerrutores.
Palavras chaves
DFT; Porfirinas; TD-DFT
Introdução
Interruptores moleculares são substâncias que se interconvertem entre estados
estáveis com propriedades distintas. A conversão entre esses estados ocorre como
resposta a diferentes estímulos, como mudança de pH, solvente, temperatura, e
interação com a luz. Quando essa interconversão entre os isômeros advém de um
estímulo luminoso, o interruptor recebe o nome de fotointerruptor(WANG, G.;
ZHANG, J., p. 299, 2012). Em 2018, Canfield e colaboradores descobriram um novo
tipo fundamental de
isomeria em compostos porfirínicos baseados na inversão de ângulo de uma ligação
simples (IAL), denominado akamptisomeria (CANFIELD et al, p. 615, 2018). O
processo de isomerização é caracterizado pela IAL, possuindo uma barreira
energética suficientemente alta (> 22 Kcal mol–1) para que seja
possível isolar
seus isômeros. Durante esse processo, os anéis pirrólicos sofrem distorções e a
planaridade do sistema é afetada, o que pode influenciar a conjugação dos
elétrons π e, consequentemente, as propriedades eletrônicas do sistema. Como
compostos porfirínicos apresentam conhecida interação com a radiação
eletromagnética, a alteração em sua estrutura eletrônica no processo de
akamptisomerização os tornam possíveis fotointerruptores moleculares. Por se
tratar de uma descoberta recente, apenas uma referência sobre o tema foi
encontrada na base de dados Scopus, além do trabalho seminal de Canfield, onde o
fenômeno de akamptisomeria frente a processos de transferência eletrônica foi
explorado para compostos com o fulereno. (STASYUK, A. J. et al, p. 2577, 2019)
Dessa forma, é crucial o aprofundamento acerca dos requisitos estruturais para a
existência de akamptisomeros isoláveis e possível aplicação em tecnologias
fotoresponsivas. Com base nisso, o presente trabalho objetiva avaliar
computacionalmente como modificações estruturais no macrociclo porfirínico
influenciam o fenômeno de akamptisomerização e suas respectivas propriedades
ópticas, idealizando a construção de fotointerruptores moleculares.O fenômeno de
inversão do ângulo de ligação da ponte ancorada (N)2 FB–O–
BF(N)
2 foi avaliado para diversos compostos. Para tanto, foram
selecionados substituintes aromáticos para avaliar a influência da extensão da
conjugação eletrônica a partir da inserção dos grupamentos na posição β,β-
pirrólica (Figura 1, Q1). Além desses, grupos doadores (Figura
1, 2a-e) e retiradores de densidade eletrônica (Figura 1, 2f-h)
foram explorados visando identificar dispositivos do tipo push-pull
(Q2). Tais sistemas permitem transferência intramolecular de
carga, uma vez que em um lado da molécula é anexado um grupamento doador de
elétrons enquanto do outro existe um grupo aceptor. (BURES, F., p. 58826, 2014)
Material e métodos
O software Gaussian 09 foi utilizado para a execução de todos os cálculos
baseados em mecânica quântica. As estruturas foram otimizadas utilizando a
Teoria do Funcional de Densidade (DFT), em nível B3LYP/6-31+G(d,p). O emprego de
tal funcional se dá pelo fato da aplicação do mesmo em compostos similares
encontrados na literatura. (CANFIELD et al, p. 616, 2018) Cálculos de
frequências vibracionais foram realizados para caracterizar a natureza dos
pontos estacionários e obter parâmetros termodinâmicos, calculados a partir de
equações clássicas da termodinâmica estatística. As energias eletrônicas foram
refinadas a partir de cálculos de ponto único utilizando o conjunto de função de
base def2-QZVP. Os espectros eletromagnéticos foram simulados utilizando a
Teoria do Funcional de Densidade Dependente do tempo (TD-DFT) em nível CAM-
B3LYP/6-31+G(d,p). Tal funcional separado por alcance foi selecionado por
apresentar uma maior eficiência na descrição de processos de transferência de
cargas, (DRAZEWIECKA-MATUSZEK, A.; RUTKOWSKA-ZBIK, D., p. 7176, 2021) sendo
muito utilizado no estudo de compostos porfirínicos. (STASYUK, A. J. et al, p.
2577, 2019) Espectros teóricos foram obtidos a partir de transições eletrônicas
verticais singleto-singleto utilizando 80 estados excitados de cada sistema
(nstates=80, singlets). Dessa forma foi possível avaliar a interação das
estruturas com a luz a partir de cálculos de excitação eletrônica, para a
avaliação das transições de maiores contribuições e a construção dos espectros
no UV/Vis.
Resultado e discussão
Inicialmente, a estabilidade relativa dos isômeros foi avaliada a partir dos
pares de akamptisômeros na configuração cisoide (c) e
transoide (t) para cada sistema (Figura 2). As
estruturas
na configuração cisoide, em que os átomos de flúor ligados aos átomos de
boro encontram-se para o mesmo lado, apresentaram maior energia (~ 6 Kcal
mol–1) do que seus equivalentes transoides. Isso possivelmente
se relaciona com a maior tensão da ponte B-O-B e elevada repulsão 1,3-
diaxial
dos átomos de flúor (Figura 2a). Além disso, não foi possível otimizar os
akamptisômeros cisoide (c2) para nenhum dos
sistemas explorados, uma vez que tal configuração não representa um ponto mínimo
de energia na superfície de energia potencial, de modo que durante o processo de
otimização, tal estrutura converge para seu par mais estável (
c1) – Figura 2a, resultado que concorda com estudos
previamente feitos. (CANFIELD et al, p. 616, 2018) A obtenção de
c2 como ponto de mínimo da superfície de energia potencial
foi apontando apenas para compostos com substituição dos átomos de flúor por um
grupo alquinil conectado ao fulereno. (STASYUK, A. J. et al, p. 2577, 2019)
Para os compostos aqui avaliados (ponte (N)2FB–O–BF(N)
2),
o par de akamptisômeros transoides se revelaram como as estruturas mais
estáveis. Segundo os resultados obtidos para os cálculos das barreiras
associadas ao IAL, todos os sistemas (t) avaliados se tratam de
espécies isoláveis. O valor médio calculado para as IAL de cada par
akamptisomerico é de ΔG‡ = 26,8 ± 0,8 Kcal mol–1(Figura
2b), ultrapassando o mínimo estabelecido como 22 Kcal mol–1 para
que
o fenômeno de akamptisomeria aconteça. (CANFIELD et al, p. 616, 2018) Em relação
aos processos competitivos à IAL, as rotações simultâneas das ligações (BF)−O−
(BF) e a dissociação do flúor possuem barreiras calculadas para sistemas
análogos em torno de 50 Kcal mol-1 (CANFIELD et al, p. 616, 2018),
impedindo a ocorrência desses processos nas condições experimentadas. Portanto,
a inferioridade da barreira relacionada à inversão do ângulo de ligação em
relação aos processos competitivos torna a akamptisomerização o processo
preferencial.
Em seguida, avaliou-se a interação das espécies akamptisoméricas com a radiação
eletromagnética a partir de cálculos TD-DFT e simulação do espectro de absorção.
Além das espécies supramencionadas, o espectro de absorção do sistema com
substituição Q = H foi calculado visando ter um referencial para
comparar o deslocamento provocado pelos substituintes explorados. Dentre as
espécies avaliadas, o maior deslocamento batocrômico foi identificado para
akamptisômeros com substituintes aromáticos, com Δλ e 21, 30 e 54 nm para os
sistemas 1a, 1b e 1c, respectivamente, com influência mais marcante
pertencendo a naftoquinona foi identificado para o sistema da naftoquinona
(1c). Isso sugere que a extensão da cadeia conjugada apresenta maior
efeito no perfil de absorção do que espécies com pares de elétrons n, que
apresentaram Δλ < 11 nm. Contudo, os constituintes de cada par de isômeros
apresentaram absorção em regiões muito próximas do espectro eletromagnético. Tal
fator indica que somente uma substituição na posição β,β-pirrólica não foi capaz
de enquadrar os sistemas como candidatos a fotointerruptores, por mais que
exista o fenômeno de IAL.
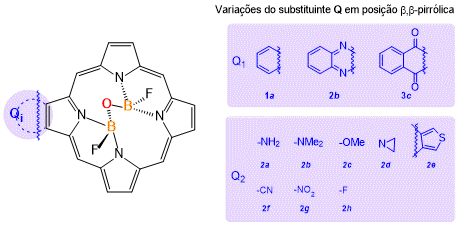
Estrutural geral de porfirinas com ponte (N)2FB-O- BF(N)2 e substituições na posição Q (β,β-pirrólica).
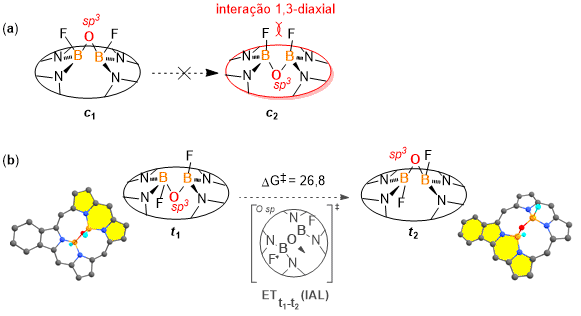
(a) Pares de akamptisômeros cisóides. Estrutura c2 não foi encontrada (converge para c1). (b) Processo de inversão do ângulo de ligação t1 → t2.
Conclusões
Segundo os cálculos apresentados, todos compostos avaliados representam espécies
akamptisoméricas isoláveis, uma vez que as barreiras energéticas para a IAL
ultrapassaram o valor mínimo estabelecido (22 Kcal mol -1). Acerca das
propriedades ópticas, identificou-se que a inserção do substituinte naftoquinona
promove um pronunciado deslocamento batocrômico se comparado com o sistema
referencial sem substituintes. Entretanto, uma única substituição na posição
Q não é capaz de enquadrar os sistemas como fotointerruptores, pois os
isômeros apresentam o mesmo perfil de absorção. A partir de tais conclusões, novas
modificações estruturais estão sendo exploradas, como saturação da substituição na
posição Q, inserção de substituintes na posição meso e avaliação de outros
substituintes aromáticos, como BODIPY e quinolonas.
Agradecimentos
Aos órgãos de fomento FAPERJ, CNPq e supercomputador LoboC – NACAD.
Referências
BURES, F.. Fundamental aspects of property tuning in push-pull molecules. RSC Adv. v. 4 , p. 58826-58851, 2014.
CANFIELD, P. J et al. A new fundamental type of conformational isomerism. Nature Chemistry.v. 10, p. 615-624, Jun. 2018.
DRAZEWIECKA-MATUSZEK, A.; RUTKOWSKA-ZBIK, D., Application of TD-DFT Theory to Studying Porphyrinoid-Based Photosensitizers for Photodynamic Therapy: A Review. Molecules, v. 26, p. 7176, Nov. 2021
STASYUK, A. J. et al, Peculiar Photoinduced Electron Transfer in Porphyrin–Fullerene Akamptisomers. Chem. Eur. J., v. 25., p. 2577 – 2585, 2019.
WANG, G. ; ZHANG, J. Photoresponsive Molecular Switches for Biotechnology. Journal of Photochemistry and Photobiology. C: Photochemistry Reviews. v. 43, n. 4, p. 299-309, Dez. 2012














