Autores
Rocha, P.L. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Júnior, F.M.S. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Sérgio, P. (UNIVERSIDADE FEDERAL FLUMINENSE) ; Fiorot, R.G. (UNIVERSIDADE FEDERAL FLUMINENSE)
Resumo
2-Aril-2,3-diidro-4(1H)-quinolonas são compostos sintéticos de importante
aplicação antitumoral. Apesar de prontamente sintetizados como mistura racêmica
através da reação one-pot de 2-aminoacetofenona com vários aldeídos aromáticos, a
atividade antitumoral até vinte vezes maior dos isômeros S da classe de
compostos os torna um objeto de interesse da síntese assimétrica. No presente
trabalho, realizou-se a avaliação in silico de um rol de oito catalisadores
quirais baseados em catalisadores de MacMillan através de cálculos de mecânica
quântica. Apresentou-se dois modelos estereopreditivos, assim como o estudo
sistêmico da importância de interações não-covalentes e da trajetória de Bürgi-
Dunitz para a origem da enantiosseletividade na reação.
Palavras chaves
imidazolidinonas; dft; mannich intramolecular
Introdução
2-Aril-2,3-diidro-4(1H)-quinolonas são compostos sintéticos análogos às flavanonas e importantes no
desenvolvimento de fármacos devido às suas propriedades antitumorais. A síntese assimétrica desses compostos é
objeto de interesse da comunidade científica, uma vez que os enantiômeros S são até 20 vezes mais
potentes na capacidade de inibição da polimerização tubulina em relação à sua mistura racêmica (PINHEIRO et
al, p. 400-406, 2017) Entretanto, a síntese one-pot mediante reação da 2-aminoacetofenona (1) e
benzaldeído (2) gerando 2-aril-2,3-diidro-4(1H)-quinolonas (3a-b) notavelmente gera um
racemato — o que torna desejável o desenvolvimento de um organocatalisador quiral como solução mais verde e
econômica para a obtenção da classe de compostos (BHEEMANAPALI et al, p. 1741-1750, 2012). Entre os
avanços recentes na área, destaca-se o desenvolvimento bem-sucedido de Wang e colaboradores de um
organocatalisador quiral aromático para a produção do enantiômero R das 2-aril-2,3-diidro-4(1H)-
quinolonas em alto rendimento e pureza óptica, além da análise de espectroscopia RMN da mistura reacional —
que revelou a formação de um intermediário imina no percurso da reação, em acordo com a proposta de que a
reação de 1 e 2 sob controle cinético consistiria numa reação de Mannich intramolecular
assimétrica (WANG et al, p. 25375-25378, 2016). Motivados pelo sucesso de Wang e colaboradores, o
presente trabalho realizou a avaliação in silico de oito organocatalisadores quirais baseados em
imidazolinonas catalisadores de Macmillan. Realizou-se para isso cálculos de mecânica quântica empregando
teoria do funcional de densidade (DFT). Estimou-se o excesso enantiomérico para cada catalisador a partir da
modelagem dos estados de transição previstos para a etapa decisória da estereoquímica dos produtos da reação
de Mannich intramolecular.
Material e métodos
Sob controle cinético, estima-se o excesso enantiomérico a partir da diferença entre
as energias de ativação entre os estados de transição levando a cada um dos
produtos. Entretanto, dado que um mesmo precursor pode levar a dois produtos óticos
distintos num caminho de reação diastereoisomérico, determina-se a diferença entre as
energias de ativação como puramente a diferença entre as energias livres de Gibbs a
temperatura constante dos estados de transição (TS) menos energéticos levando a cada
um dos produtos óticos possíveis. Devido ao grande número de geometrias avaliadas
para cada sistema, utilizou-se uma comum abordagem de correção da energia eletrônica
(GOODMAN, M. J.; SIMÓN, L., p. 689-700, 2018). Para estimar a diferença entre as
energias de ativação realizou-se uma abordagem , empregando cálculos de teoria do
funcional de densidade (DFT) no nível de teoria B3LYP/6-31G(d) para a otimização das
geometrias de TS e caracterização via cálculo de frequência vibracional e, para
melhor caracterização da energia eletrônica das geometrias dos TSs, realizou-se o
cálculo de single point no nível de teoria M06-2X/6-311+G(d,p) e somou-se ela à
correção térmica à energia livre de Gibbs obtido via cálculo de frequência no nível
de teoria B3LYP/6-31G(d). Todos os cálculos foram realizados no pacote Gaussian09.
Resultado e discussão
Dada a ausência de unidade ácida acessível no rol de catalisadores propostos,
dois mecanismos foram estudados. No primeiro, ocorreria a formação de um
intermediário íon imínio precedendo a formação da ligação C—C na etapa decisiva
da estereoquímica dos produtos. No segundo, não há ativação da imina no ciclo
catalítico. Para o modelo prevendo a formação de um intermediário íon imínio,
sugere-se que a ativação da carbonila pelo catalizador precedendo a formação do
intermediário enamina liberaria prótons na mistura reacional — que uma vez
removidos pelo solvente, poderiam protonar a imina. Assumindo o intermediário
imínio como precursor, observou-se uma importante contribuição de interações
aromáticas NH-pi e CH-pi entre o imínio na estabilização das geometrias de TS
Pro-S e Pro-R menos energéticas. Semelhantemente a trabalho
prévios onde interações aromáticas se mostraram decisivas para o controle da
enantioseletividade (KRENSKE, H. E, p. 5226-5232, 2013) verificou-se que
substituir o grupamento fenila aromático do catalisador por uma variedade de
grupamentos que desfavorecem ou inibem as interações NH-pi e CH-pi entre o
imínio e a unidade aromática do catalisador de fato levaram a uma redução da
performance esperada do catalisador traduzida por menor ee% a favor do
desejado produto S. Concomitantemente, a substituição do grupamento
fenila aromático por outro benzila levou a geometrias mais estericamente
impeditivas para a estabilização da formação da ligação C-C através da face
Si (Pro-S) por interações aromáticas do tipo NH-pi, observadas
como decisivas para o controle da enantiosseletividade no sistema e também em
bom acordo com a literatura para sistemas semelhantes. (KRENSKE, H. E.; HOUK,
K.N., p. 979-989, 2013). Já para o mecanismo tomando o intermediário imina como
precursor da etapa decisória da estereoquímica dos produtos, observou-se
geometrias de TS menos energéticas como aquelas contendo ângulos mais largos de
Burgi-Dunitz e por isso menores distâncias C-C nos estados de transição da etapa
decisória da estereoquímica dos produtos, além de ausência de interações de
interações aromáticas nestas. Sugeriu-se então que o controle da
enantiosseletividade no modelo estereopreditivo se daria em maior caráter
através da minimização da repulsão estérea entre o catalisador e o restante da
molécula catalisada. De fato, assumindo-se a imina como intermediário, obteve-se
um razoável fator de correlação linear (R2 = 0.75), em bom acordo com
o esperado pela literatura para a reação de Mannich catalisada por L-prolina.
(SURESH, H.C.; AJITHA, J. M., p. 1962-1970, 2011). Ambos os modelos
estereopreditivos estão descritos na Fig. 1. Apesar do controle da
enantiosseletividade ter origens distintas em ambos os modelos
estereopreditivos, estimou-se performance similar (97% ee) para os
catalisadores de melhor performance P1 e P2 assumindo
tanto o íon imínio como a imina como precursores. Registrou-se o rol de
catalisadores estudados e a performance esperada na Tabela 1.
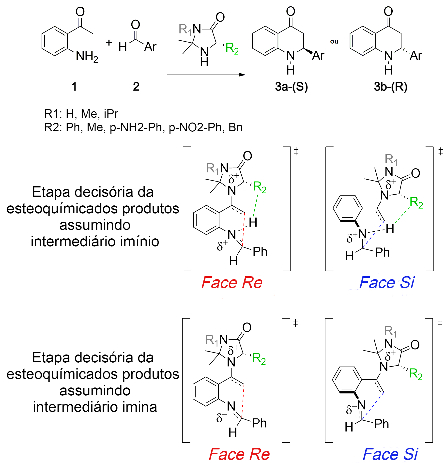
Rol de catalisadores avaliados e modelos estereopreditivos dos estados de transição para a etapa decisória da estereoquímica dos produtos
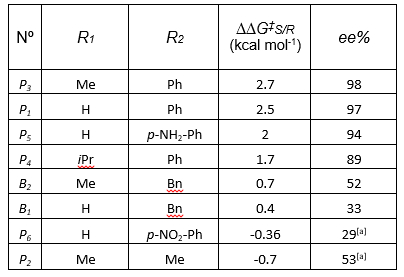
Relação de catalisadores e performance esperada assumindo intermediário íon imínio
Conclusões
O rol de catalisadores testados se mostrou promissor e os modelos
estereopreditivos propostos oferecem uma plataforma mais rápida para a avaliação
in silico da reação de 2-aminoacetofenona e benzaldeído para uma maior
variedade de aldeídos aromáticos de interesse farmacológico.
Agradecimentos
Capes, Faperj e CNPq.
Referências
BHEEMANAPALI, L. N.; KAUR, A.; SANGEETA, R.; JAVALI, N. M. Synthesis, evaluation of 6,8-dibromo-2-aryl-2,3-dihydroquinolin-4(1H)-ones in MCF-7 (breast cancer) cell lines and their docking studies. Medical Chemical Research, no. 72, 1741-1750, 2012.
GOODMAN, M. J.; SIMÓN, L. How reliable are DFT transition structures? Comparison of GGA, hybrid-meta-GGA and meta-GG funcionals. Organic Biomolecular Chemistry, no. 9, 689-700, 2018.
KRENSKE, H. E.; HOUK, K. N. Aromatic interactions as control elements in stereoselective organic reactions. Accounts of chemical research, no. 4, 979-989, 2013.
KRENSKE, H. E.; Aromatic interactions as control elements in stereoselective organic reactions. Organic biomolecular chemistry, no. 4, 5226-5232, 2013.
PINHEIRO, S.; MURI, E. M. F.; OLIVEIRA, R. P. R. F; DIAS, L. R. S.; GRECO S. J. Asymmetric catalysis in the synthesis of azaflavanones. Mini Reviews in Organic Chemistry, no. 5, 400-4006, 2017.
SURESH, H.C.; AJITHA, J.M. Role of stereoelectronic features of imine and enamine in (S)-proline catalyzed Mannich reaction of acetaldehyde: an in silico study. Journal of Computational Chemistry, no. 9, 1962-1970, 2011.
WANG, Y. Q.; PAN, G. F.; SU, L.; ZHANG, Y. L.; GUO, S. H. Organocatalytic one-pot asymmetric synthesis of 2-aryl-2,3-dihydro-4-quinolones. Royal Society of Chemistry Advances Advances, no. 6, 25375-25378, 2016.














