Autores
Silva, L.G.P. (UEMA) ; Serra, K.F.C. (UEMA) ; Gomes, T.F. (UEMA) ; Ferreira, L.A. (UEMA) ; Gonçalves, J.C.S. (UEMA) ; Oliveira, A.L.T. (UEMA) ; Santos, P.L.L. (UEMA) ; Fernandes, R.M.T. (UEMA) ; Khan, A. (UEMA)
Resumo
Os métodos computacionais vêm sendo empregados na química como investigadores de
propriedades físico-químicas e vibracionais de isômeros de substâncias já
conhecidas e que podem servir de protótipos para combustíveis, dominar a
estrutura que se trabalha é essencial, pois a partir de suas descrições
energéticas e estruturais, pode-se analisar a interação da substância com seu
alvo. A metodologia empregada foi a modelagem molecular feita pelos programas
Gaussview e Gaussian 9, onde foram obedecidas as distâncias entre as ligações de
C-C, C-H, C-O e O-H para cada isômero modelado. As otimizações foram trabalhadas
com o modelo funcional B3LYP, PBE0 e PM6 de DFT, utilizando um conjunto de base
cc-pVTZ. Já para o cálculo de frequências foram utilizado os conjuntos de base
B3LYP, PB30 e PM6.
Palavras chaves
HOMO-LUMO; DFT; GAP DE ENERGIA
Introdução
O chamado gap de energia entre as regiões HOMO-LUMO das moléculas é um ramo da
química quântica. A teoria dos orbitais moleculares (OMs) diz que uma
determinada moléculas tem seus orbitais formados através da combinação linear de
orbitais anteriormente atômicos. É neste ambiente de orbitais de fronteiras,
especialmente o HOMO e o LUMO, que ocorrem a interação entre as moléculas e são
responsáveis por muitas das características químicas e físicas das mesmas.
Quando menor a diferença, mais próximos estarão os níveis de energia HOMO e
LUMO, menor será a barreira energética e mais provável dá-se a passagem de
elétrons do HOMO para o LUMO (ROCHA, 2005).
Com a sobreposição de orbitais moleculares sobre outros orbitais criam uma
interação eletrônica de novos orbitais, os chamados ligantes (mais baixa
energia) e os antiligantes (mais alta energia). A diferença de energias nesses
orbitais HOMO e LUMO cria a estabilidade e o ambiente reativo das moléculas, e
esta reatividade se calcula com o gap HOMO-LUMO. Quando há uma alta estabilidade
nesse intervalo de orbitais, haverá baixa reatividade química e pouca formação
de novos compostos.
O objetivo deste trabalho é investigar as interações eletrônicas e vibracionais
das moléculas ditas isômeros do butanol, e a partir desse ponto poder afirmar ou
predizer se são passíveis de aplicação na octanagem da gasolina.
Material e métodos
Neste estudo, primeiramente foi realizado o cálculo e modelagem de geometria e
posteriormente foi feita a frequência, foi utilizado o funcional híbrido de 3
parâmetros de Becke combinado com o funcional gradiente-correlação de Lee–Yang–
Parr (B3LYP) e o funcional puro de Perdew, Burke e Ernzerhof como um híbrido por
Adamo (PBE0) (ADAMO; BARONE, 1999) foram testados nos cálculos usando a Teoria
do Funcional da Densidade (DFT). Todos os conjuntos de bases polarizadas (cc-
pVTZ) consistentes com correlação de dunnings de elétrons (zeta triplo) foram
empregados. Um método semi-empírico no hamiltoniano PM6 (STEWART, 2007) também
foi empregado, conforme implementado em gaussiano.
A partir dos resultados obtidos foi feita a própria produção de energias de HOMO
e LUMO no programa, que mostrou os seus resultados dos três funcionais. Com
isso, foi observado como há as interações das estruturas de aditivos (N-butanol,
Sec-butanol, Terc-butanol e Iso-butanol) com seus alvos e quais podem apresentar
melhores resultados para serem utilizados em propelentes.
Resultado e discussão
O cálculo das moléculas apresentou os dados bem distintos para cada método
utilizado. Segundo a tabela 1, quem apresentou o Gap de energia mais baixo foi o
Terc-butanol na base PBE0, com o valor de 0,22393. A partir desse ponto
percebeu-se um paralelo entre todos os cálculos que foi sempre da metodologia
PBE0 ser a de menor valor entre todos os cálculos, isso vai ao encontro de como
é feita essa metodologia no programa Gaussian 9, quem distingue bem os valores
dos cálculos.
Outro ponto importante foi a pouca interação (alto Gap) em relação aos valores
encontrados na metodologia de PM6, isso por que as densidades modeladas não se
aproximaram tanto e os cálculos tornaram-se diferentes das outras metodologias
usadas. Pode-se afirmar que os métodos semiempíricos buscam encontrar soluções
com proximidade a equação de Schrodinger e empregam parâmetros ditos empíricos e
algumas restrições matemáticas mais pontuais do que as utilizadas em métodos ab
initio (LASCHUK, 2005), nesse ponto pode haver diferença nos resultados
encontrados.
Com estes resultados, podemos afirmar que os menores valores podem identificar
uma menor barreira na passagem dos elétrons dos orbitais homo para lumo e com
isso uma alta reatividade, inferindo assim numa boa reação química das moléculas
com outros reagentes que podem ser utilizados posteriormente em estudos de
aditivos da gasolina. Isso é um bom resultado para se saber uma predição sem a
utilização dos reagentes em questão, ajudando além de estudos preliminares a
diminuição de compostos no meio ambiente, grande ponto para a utilização da
química computacional.
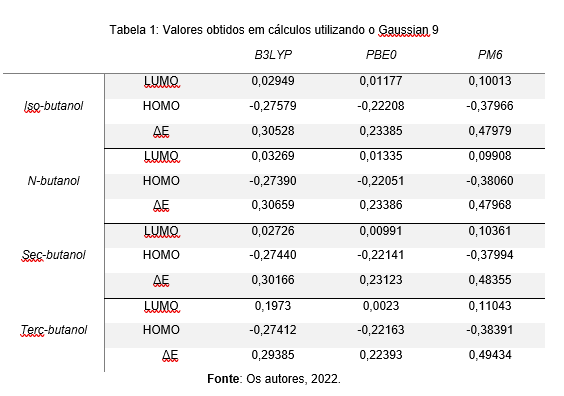
Tabela 1: Valores obtidos em cálculos utilizando o Gaussian 9
Conclusões
Concluiu-se que as moléculas apontam como precursoras na abordagem da modelagem
molecular e suas interações vibracionais e eletrônicas quanto para possível aplicação
prática como aditivos de gasolina. Alcançou-se os objetivos, pois podemos observar os
valores de interações entre os orbitais moleculares (OMs) e como podem servir de
elucidação para possíveis novas dúvidas na química computacional.
Agradecimentos
Ao meu orientador Dr. Alamgir Khan e ao meu grupo do Laboratório de Físico-química da
Universidade Estadual do Maranhão – UEMA. Ao suporte de bolsa FAPEMA durante os
últimos anos de fomento.
Referências
ADAMO, Carlo; BARONE, Vincenzo. Toward reliable density functional methods without adjustable parameters: The PBE0 model. The Journal of chemical physics, v. 110, n. 13, p. 6158-6170, 1999.
DUNNING, T. H.; PETERSON, K. A.; WOON, D. W.; SCHLEYER, P. V. R. “Encyclopedia of Computational Chemistry”, Vol. 1, Wiley, New York, 1998, p. 88-115.
LASCHUK, E. F. Novo Formalismo Semiempírico para Cálculos Químicos-Quânticos. Tese. Universidade Federal do Rio Grande do Sul. Programa de Pós-graduação em química. 2005. Disponível em:<https://www.lume.ufrgs.br/bitstream/handle/10183/7140/000495676.pdf?sequence=1&isAllowed=y>.
MARINHO, Márcia Machado; CASTRO, Rondinelle Ribeiro; MARINHO, Emmanuel Silva. Utilização do método semi-empírico PM7 para caracterização do fármaco atalureno: HOMO, LUMO, MESP. Revista Expressão Católica Saúde, v. 1, n. 1, 2016.
Stewart, James J. P. "Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements." Journal of Molecular Modeling, v. 13, n. 12, 2007, 1173-213. Disponível em: DOI: 10.1007/s00894-007-0233-4.














