TÍTULO: Utilização da Química Computacional para o Ensino de Reações Químicas: Reações de Halogenação
AUTORES: CORDEIRO, D. O. (UFCG) ; SANTANA, O. L. DE (UFCG)
RESUMO: Este trabalho tem como objetivo criar um banco de dados de reações de halogenação para a elaboração de material didático que auxilie no ensino de importantes conceitos em Química. Utilizou-se o GaussView 3.0 e o Gaussian 03 como programas de modelagem molecular e visualização estrutural. Os dados foram obtidos pelo método DFT, com o funcional B3LYP e o conjunto de base 6-31+G*, com o objetivo de descrever sistemas carregados, bem como a quebra e a formação de ligações no estado gasoso. Os resultados qualitativos para os sistemas em fase gasosa referentes as geometrias otimizadas são consistentes com o mecanismo iônico (de duas etapas).
PALAVRAS CHAVES: ensino de química, química computacional, mecanismo de reação.
INTRODUÇÃO: Diante da preocupação crescente de como transmitir determinadas informações teóricas aos alunos, este trabalho visa à inclusão de avanços tecnológicos recentes no aprimoramento do processo de ensino-aprendizagem [1]. Neste trabalho, alguns exemplos de reações de halogenação são investigados.
Os alcenos reagem com moléculas de halogênio X2 (mais especificamente, com cloro e bromo) em solventes não-nucleofílicos para formar dialetos vicinais. Em solventes não-nucleofílicos, a reação eletrofílica segue um mecanismo iônico de duas etapas. Na primeira, os elétrons da ligação π do alceno atacam a molécula de halogênio X2 à medida que esta se aproxima da dupla ligação, polarizando-a e fazendo com que o halogênio (1)X, mais próximo à dupla ligação, atue como um eletrófilo. Desta forma a ligação no halogênio é quebrada, formando um ânion (2)X e um cátion (1)X+, que recebe um par de elétrons da dupla ligação, estabilizando a carga positiva por deslocalização e resultando em um cátion de cadeia fechada de três membros. Na segunda etapa, o ânion (2)X-, produzido na etapa anterior, ataca o carbono (2)C, resultando na formação de um dialeto vicinal através da abertura do anel de três membros [2].
O objetivo deste trabalho é a criação de um banco de dados gerado por métodos de modelagem computacional, de modo que o estudante possa visualizar o comportamento molecular em diferentes aspectos. Investigou-se a reação não-catalisada em fase gasosa, de modo a possibilitar o estudo de um mecanismo concertado em uma única etapa, considerando apenas um substituinte, com X = Cl e R = H, F, Cl, OH, NH2 e CH3.
MATERIAL E MÉTODOS: Este trabalho foi desenvolvido em um computador pessoal com a seguinte configuração: processador Celeron D 3.06 GHz, com 512 Mb de RAM e HD 80 Gb. Utilizou-se o GaussView 3.0 [3] e o Gaussian 03 [4] como programas de visualização estrutural e modelagem molecular.
Os mecanismos das reações investigadas neste trabalho foram obtidos pelo método DFT [5] (método pseudo-correlacionado), com a configuração B3LYP/6-31+G* [6-8], a partir das seguintes etapas: (i) otimização completa das geometrias dos reagentes e produtos seguida por cálculo de freqüência; (ii) busca pelo estado de transição, utilizando a metodologia STQN [9-10]; (iii) otimização completa do estado de transição seguida por cálculo de freqüência; e (iv) determinação da coordenada de reação a partir da implementação IRC (Intrinsic Reaction Coordinate) [11-12]. Todas as etapas foram executadas utilizando-se o programa GausView para a preparação das estruturas relativas a reagentes, produtos e estados de transição, de acordo com o descrito em trabalhos anteriores [1]. Todas as otimizações de geometria foram seguidas por um cálculo de freqüências, com o objetivo de confirmar que as estruturas obtidas correspondem a uma estrutura de estado estacionário, de mínimo (reagentes e produtos) ou de máximo de energia (estado de transição): a inexistência de freqüências imaginárias para reagentes e produtos confirma que estas correspondem a estruturas de mínimo de energia; um única freqüência imaginária para o estado de transição confirma que a geometria obtida corresponde a uma estrutura legítima de máximo de energia. O cálculo da coordenada de reação foi realizado para confirmar se a geometria obtida para o estado de transição corresponde a uma estrutura correta, consistente com o mecanismo proposto.
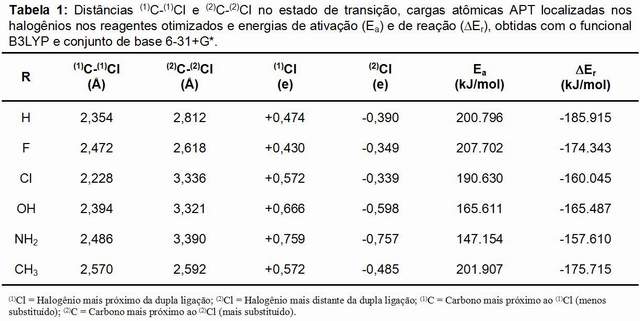
RESULTADOS E DISCUSSÃO: Os principais resultados obtidos neste trabalho correspondem às propriedades estruturais (geometrias) e eletrônicas (cargas atômicas e energias relativas). As geometrias otimizadas não apresentam mudanças significativas entre as diferentes estruturas dos reagentes, produtos e estados de transição para os diferentes substituintes considerados em relação ao sistema de referência com R = H, possibilitando a interpretação dos resultados a partir da consideração de um único mecanismo.
Embora a reação investigada siga um mecanismo concertado (em uma única etapa), os resultados obtidos são consistentes com o mecanismo iônico (de duas etapas), que segue a generalização da chamada regra de Markovnikov [2]. Nas reações investigadas, o eletrófilo corresponde ao halogênio (1)Cl, mais próximo da dupla ligação. De acordo com a Tabela 1, os estados de transição determinados apresentam a característica de que o halogênio (1)Cl (o eletrófilo) ataca primeiro o carbono (1)C (carbono mais hidrogenado), o que é indicado pela menor distância (1)C-(1)Cl.
O cálculo de estrutura eletrônica possibilita determinar, dentre outras propriedades moleculares, as cargas atômicas parciais localizadas. Neste trabalho foram determinadas as cargas APT (do inglês Atomic Polar Tensor) [13], mostradas na Tabela 1. O resultado é consistente com o mecanismo proposto, segundo o qual o halogênio (1)Cl, mais próximo à dupla ligação, apresenta carga parcial positiva, atuando como eletrófilo na reação. Em todos os casos observa-se a ocorrência de carga líquida positiva na molécula, indicando uma transferência eletrônica do halogênio Cl2 para o alceno C2H3R. As cargas parciais localizadas em (1)Cl apresentam a seguinte ordem: F < H < Cl < CH3 < OH < NH2.
CONCLUSÕES: Os resultados obtidos relativos às reações de halogenação permitem concluir que:
A interpretação dos resultados, a partir da consideração de um único mecanismo concertado, foi possibilitada pela determinação de estruturas relativas a reagentes, produtos e estados de transição similares para os diferentes substituintes considerados, em relação ao sistema de referência com R = H.
Embora a reação investigada siga um mecanismo concertado (em uma única etapa), os resultados obtidos são consistentes com o mecanismo iônico (de duas etapas), seguindo a generalização da regra de Markovnikov.
AGRADECIMENTOS: Ao CNPq pela bolsa de Iniciação Científica PIBIC/UFCG/CNPq.
REFERÊNCIAS BIBLIOGRÁFICA: 1.A. Mariano, E. V. Monte, S. A. Monte, C. F. Braga, A. B. Carvalho, R. C. M. U. Araújo, O. L. Santana; O Ensino de Reações Orgânicas Usando Química Computacional: I. Reações de Adição Eletrofílica a Alquenos; Quím. Nova 31(5) (2008) 1243.
2.P. Y. Bruice; Química Orgânica, Ed. Pearson, 4a Edição (2006).
3.GaussView 3.0; Gaussian: Pittsburgh, PA (2004).
4.Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann Jr., R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Salavador, P.; Dannenberg, J. J.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin; R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A; Gaussian 03, Revision B.02, Gaussian, Inc., Pittsburgh, P. A. (2003).
5.Hohenberg, P.; Kohn, W.; Phys. Rev. B 136 (1964) 864.
6.Becke A. D.; J. Chem. Phys. 98 (1993) 5648.
7.Hehre, W. J.; Ditchfield, R.; Pople, J.A.; J. Chem. Phys. 56 (1972) 2257.
8.Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J.A.; J. Chem. Phys. 72 (1980) 650.
9.Peng, C. Y.; Schlegel, H. B.; Isr. J. Chem. 33 (1993) 449.
10.Peng, C. Y.; Ayala, P. Y.; Schlegel, H. B.; Frisch, M. J.; J. Comp. Chem. 17 (1996) 49.
11.Gonzalez, C.; Schlegel, H. B.; J. Chem. Phys. 90 (1989) 2154.
12.Gonzalez, C.; Schlegel, H. B; J. Chem. Phys. 94 (1990) 5523.
13.Cioslowski, J.; J. Am. Chem. Soc. 111(22) (1989) 8333.
