
Realizado em Teresina/PI, de 28 a 30 de Julho de 2013.
ISBN: 978-85-85905-05-7
TÍTULO: A ACIDEZ DE AMINOÁCIDOS ANALISADA ATRAVÉS DE MÉTODOS COMPUTACIONAIS
AUTORES: Moura, L.C.B. (UFPI) ; Alencar, H.A.C. (UFPI) ; Moita Neto, J.M. (UFPI)
RESUMO: Os valores de pKa dos 20 α-aminoácidos foram estudados teoricamente empregando
métodos de cálculos ab initio. O objetivo foi mostrar a influência da
estrutura eletrônica das moléculas no comportamento da acidez desses aminoácidos.
Parâmetros de cargas foram analisados a fim de correlacioná-los aos valores de pKa
determinados experimentalmente. A regressão linear múltipla apontou valores
aceitáveis na correlação das cargas com os valores de pKa e mostrou que
aproximadamente 30% dos valores de pKa (experimentais) estão relacionados com a
estrutura eletrônica da molécula.
PALAVRAS CHAVES: cáculo ab initio; aminoácidos; estrutura eletrônica
INTRODUÇÃO: O objetivo central da química quântica é a obtenção de soluções da equação de
Schrödinger para a determinação precisa de propriedades de sistemas atômicos e
moleculares (MORGON e COUTINHO, 2007). A dificuldade em resolver essa equação de
forma exata reforça a necessidade da utilização de métodos que se aproximem de
tal solução. Os métodos computacionais obtêm valores aproximados da equação de
Schrödinger. Pode-se dar ênfase aos métodos ab initio que se baseiam em
procedimentos de Hartree-Fock (HF) e na Teoria do Funcional de Densidade (DFT);
e métodos semi-empíricos. A utilização destes métodos depende da análise que se
deseja fazer e o tempo necessário para tal, uma vez que a demora no tempo de
cálculo (custo computacional) cresce proporcionalmente ao rigor teórico.
Os aminoácidos são subunidades monoméricas que fornecem a chave para a formação
de milhares de estruturas de proteínas diferentes que são construídas a partir
do conjunto de 20 aminoácidos que se ligam de maneira covalente em sequências
lineares características. A partir dessas inúmeras combinações, os diferentes
organismos podem fazer diferentes produtos como enzimas, hormônios, anticorpos,
antibióticos, entre outros (NELSON e COX, 2006).
Os 20 aminoácidos comuns são α-aminoácidos, eles possuem um grupo carboxila e um
grupo amina ligados ao mesmo átomo de carbono, denominado de carbono α, eles se
tornam diferentes uns dos outros nas suas cadeias laterais (NELSON e COX, 2006).
O grupo amino e o grupo carboxílico podem existir nas formas ácida ou básica,
isso depende do pH da solução em que se encontram dissolvidos (BRUICE, 2006).
O objetivo deste trabalho foi mostrar a influência da estrutura eletrônica,
fundamentada na química quântica, no comportamento da acidez dos 20 α-
aminoácidos.
MATERIAL E MÉTODOS: Cálculos computacionais foram realizados para os 20 α-aminoácidos (Ácido
aspártico, Ácido glutâmico, Alanina, Arginina, Asparagina, Cisteína, Fenil-
alanina, Glicina, Glutamina, Histidina, Isoleucina, Leucina, Lisina, Metionina,
Prolina, Serina, Tirosina, Treonina, Triptofano e Valina).
A análise mecânico-quântica foi realizada utilizando o pacote computacional
Gaussian 03W (FRISCH et al., 2003). O cálculo computacional para análise de cargas
de Mulliken foi desenvolvido utilizando o método ab initio Hartree-Fock. O
conjunto de base utilizado foi 6-31G. O software GaussView 3.0 foi aplicado para
gerar os inputs necessários para realização dos cálculos e para visualização dos
resultados obtidos.
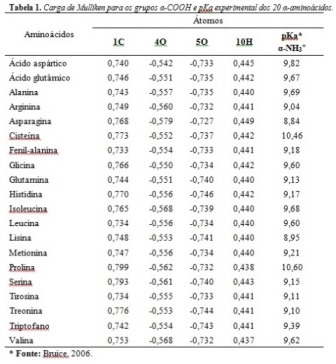
RESULTADOS E DISCUSSÃO: Os resultados dos valores de cargas dos α-aminoácidos estão apresentados na
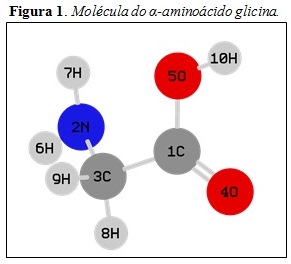
Tabela 1. A Figura 1 mostra o aminoácido glicina como exemplo para demonstração
da parte do aminoácido que foi colhida as informações de carga, representada na
Tabela 1.
Os valores de pKa são calculados a partir da determinação do pH das substâncias
experimentalmente. Alguns dos fatores que condicionam o resultado do valor de
pKa são: ressonância, eletronegatividade, tamanho do átomo, hibridização,
solvatação, carga, efeito indutivo, entre outros.
Cálculos computacionais podem ser realizados para determinação de valores de pKa
teóricos. Recursos da mecânica quântica podem ser explorados para
determinação/previsão de alguns parâmetros que são medidos experimentalmente.
Em determinações experimentais são esperadas interações molécula-molécula e
interações molécula-solvente como condicionantes do valor de pKa. No cálculo
quântico (molécula isolada no estado gasoso), não é permitido à molécula
realizar interação de qualquer natureza. Logo, não se pode prever valores de pKa
com base nos valores de carga obtidos. Entretanto, o resultado apresentado pela
regressão linear múltipla indica que aproximadamente 30% dos valores de pKa
(experimentais) estão relacionados com a estrutura eletrônica das moléculas. Ou
seja, as cargas residuais obtidas através de cálculos computacionais indicam que
a estrutura eletrônica também é um fator que influencia no valor de pKa dos
aminoácidos.
CONCLUSÕES: A análise mecânico-quântica é uma ferramenta útil na determinação de fatores que
influenciam a acidez de substâncias. Esta ferramenta pode mostrar que os valores
de pKa (experimentais) dos 20 aminoácidos pesquisados recebem uma contribuição das
cargas residuais da estrutura eletrônica dessas moléculas.
AGRADECIMENTOS:
REFERÊNCIAS BIBLIOGRÁFICA: BRUICE, P.Y. [i]Química Orgânica[/i]. 4. ed. São Paulo: Pearson Prentice Hall, 2006.
FRISCH, M. J.; TRUCKS, G. W.; SCHLEGEL, H. B.; SCUSERIA, G. E.; ROBB, M. A.; CHEESEMAN, J. R.; MONTGOMERY, JR., J. A.; VREVEN, T.; N. KUDIN, K.; BURANT, J. C.; MILLAM, J. M.; IYENGAR, S. S.; TOMASI, J.; BARONE, V.; MENNUCCI, B.; COSSI, M.; SCALMANI, G.; REGA, N.; PETERSSON, G. A.; NAKATSUJI, H.; HADA, M.; EHARA, M.; TOYOTA, K.; FUKUDA, R.; HASEGAWA, J.; ISHIDA, M.; NAKAJIMA, T.; HONDA, Y.; KITAO, O.; NAKAI, H.; KLENE, M.; LI, X.; KNOX, J. E.; HRATCHIAN, H. P.; CROSS, J. B.; ADAMO C.; JARAMILLO, J.; GOMPERTS, R.; STRATMANN, R. E.; YAZYEV, O.; AUSTIN, A. J.; CAMMI, R.; POMELLI, C.; OCHTERSKI, J. W.; AYALA, P. Y.; MOROKUMA, K.; VOTH, G. A.; SALVADOR, P.; DANNENBERG, J. J.; ZAKRZEWSKI, V. G.; DAPPRICH, S.; DANIELS, A. D.; STRAIN, M. C.; FARKAS, O.; MALICK, D. K.; RABUCK, A. D.; RAGHAVACHARI, K.; FORESMAN, J. B.; ORTIZ, J. V.; CUI, Q.; BABOUL, A. G.; CLIFFORD, S.; CIOSLOWSKI, J.; STEFANOV, B. B.; LIU, G.; LIASHENKO, A.; PISKORZ, P.; KOMAROMI, I.; MARTIN, R. L.; FOX, D. J.; KEITH, T.; AL-LAHAM, M. A.; PENG, C. Y.; NANAYAKKARA, A.; CHALLACOMBE, M.; GILL, P. M. W.; JOHNSON, B.; CHEN, W.; WONG, M. W.; GONZALEZ C.; POPLE, J. A.; [i]Gaussian 03W; Gaussian, Inc., Pittsburgh PA, 2003[/i].
MORGON, N. H.; COUTINHO, K. (Ed.). [i]Métodos de química teórica e modelagem molecular[/i]. São Paulo: Livraria da Física, 2007.
NELSON, D. L. e COX, M. M.. [i]Lehninger Princípios de Bioquímica [/i]. 4. ed. São Paulo, Sarvier, 2006.